Shan-Han Chang1, Yung-Xin Tsai1, Ta-Fu Chen2, Wan-Chen Wu1, Pei-Lung Chen3 and Jin-Ying Lu1*
1Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan
2Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan
3Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan
*Corresponding Author:
Jin-Ying Lu
Department of Internal Medicine, National
Taiwan University Hospital, Taipei 100, Taiwan
Tel: 886-2-23123456 ext. 67126
E-mail: jylu@ntu.edu.tw
Received date: November 29, 2017; Accepted date: December 06, 2017; Published date: December 11, 2017
Citation: Chang SH, Tsai YX, Chen TF, Wu WC, Chen PL, et al. (2017) A 49-Year-Old Man with Combined Myasthenia Gravis, Autoimmune Thyroiditis and IgG4 Orbitopathy. Transl Biomed. Vol.8 No.4:140 doi:10.21767/2172-0479.100140
Background: Patients with autoimmune thyroid disease have higher risk of myasthenia gravis, especially the ocular form of myasthenia gravis. IgG4-related disease and autoimmune thyroiditis have been reported to coexist; and IgG4- related thyroiditis pertain higher autoantibodies when compared with non-IgG4 thyroiditis. However, combined occurrence of myasthenia gravis, autoimmune thyroiditis, and IgG4-related disease has not been reported before.
Methods: Here we report a case of ocular myasthenia gravis combined with autoimmune thyroiditis and elevated serum IgG4 that initially deteriorated to generalized myasthenia gravis after 3 days of intravenous (IV) pulse methylprednisolone treatment, but was rescued after plasma exchange therapy. The patient was placed on oral prednisolone, azathioprine, pentoxifylline, hydroxychloroquine, but the ocular symptoms relapsed occasionally when he felt tired. Another eight sessions of weekly IV pulse methylprednisolone were administered during outpatient clinic follow-ups.
Findings: There was no more deterioration to generalized myasthenia gravis, and his ocular symptoms fully recovered after the treatment. He finally underwent thoracoscopic thymectomy, and pathology shows atrophic thymus. All immunosuppressants were tapered off after surgery, and he remained symptomfree on the last follow-up, about half a year after surgery.
Conclusion: Since thyroid associated orbitopathy and IgG4-related orbitopathy respond well to steroid treatment, initial deterioration of concomittent ocular myasthenia gravis to generalized form after IV pulse steroid therapy can be rescued by plasmapheresis, and the follow-up IV pulse steroid therapy should not be precluded as the definitive treatment in this situation. Finally, all immunosuppressants were successfully discontinued and he remained symptomfree after surgical removal of the atrophic thymus.
Keywords
Myasthenia gravis; Autoimmune thyroiditis; IgG4-related disease; Thyroid orbitopathy
Introduction
Patients with autoimmune thyroid disease have higher risk of myasthenia gravis (MG), especially ocular form of MG [1]. IgG4- related disease and autoimmune thyroid disease have been reported to coexist and pertain higher titers of autoantibodies when compared with non-IgG4 thyroiditis [2]. However, combined occurrence of MG, autoimmune thyroid disease, and IgG4-related disease has not been reported before. Here we report a case of ocular MG associated with thyroid orbitopathy and elevated IgG4 level, whose disease deteriorated to generalized MG after initiation of intravenous (IV) pulse methylprednisolone, but was rescued after plasma exchange. The ocular symptoms recurred on oral steroids and immunosuppressants, but totally recovered after additional eight sessions of IV pulse methylprednisolone therapy.
Case Report
A 49-year-old man, who had left tongue cancer post operation and radiation therapy in 2005, came to our hospital because of left upper eyelid drooping for five days. Four months before the admission, sudden onset of binocular vertical diplopia happened during jogging, associated with right eyelid ptosis. Brain magnetic resonance imaging (MRI) and electromyogram (EMG) at local hospital revealed no significant abnormal findings, but laboratory test showed high thyroid peroxidase antibody (TPOA) (>2000 IU/mL, reference range <5.6 IU/mL). The ocular symptoms gradually resolved under oral prednisolone and acupuncture. Four months later, sudden onset of blurred vision recurred after jogging, followed by left eyelid total ptosis, which did not improve after acupuncture. The ocular symptoms disappeared when he woke up, but soon recurred in the daytime. These symptoms did not hinder his activities of daily living. He sought medical advice again because of intravctable left eyelid ptosis and bilateral ophthalmoplegia. Neurological examination showed fixed eyeballs without diplopia in neutral position, left eyelid total ptosis, and mild right eyelid ptosis (Figure 1A). Repetitive stimulation test revealed no decremental change at rest and post-exercise. Extraocular movement (EOM) was absent. Ice packing test showed a negative result. Forced duction test showed freely movable eyeballs at all directions. Fundus exam disclosed no retinal lesion in both eyes. Brain CT showed proptosis of bilateral eyes without obvious extraocular muscle hypertrophy (Figure 1B). The laboratory test showed normal thyroid function, thyrotropin-binding inhibiting immunoglobulin, and lactic acid levels, but high thyroglobulin antibody (TA) (2192.27 IU/mL, reference range <14.4 IU/mL) and TPOA (>2000.00 IU/mL). Thyroid ultrasonography revealed autoimmune thyroid disease and one hypoechoic lesion in the left lower thyroid pole. His left ptosis only slightly improved after one dose of 60 mg pyridostigmine (Figure 1A). Cerebrospinal fluid study, nerve conduction velocity, F-wave, and blink reflex studies were all normal. Single fiber EMG disclosed markedly increased mean consecutive difference with conduction block, and the acetylcholine receptor autoantibody (AchRAb) was elevated (6.424 nmol/L, reference range <1 nmol/L). Chest computed tomography showed no anterior mediastinal lesion.
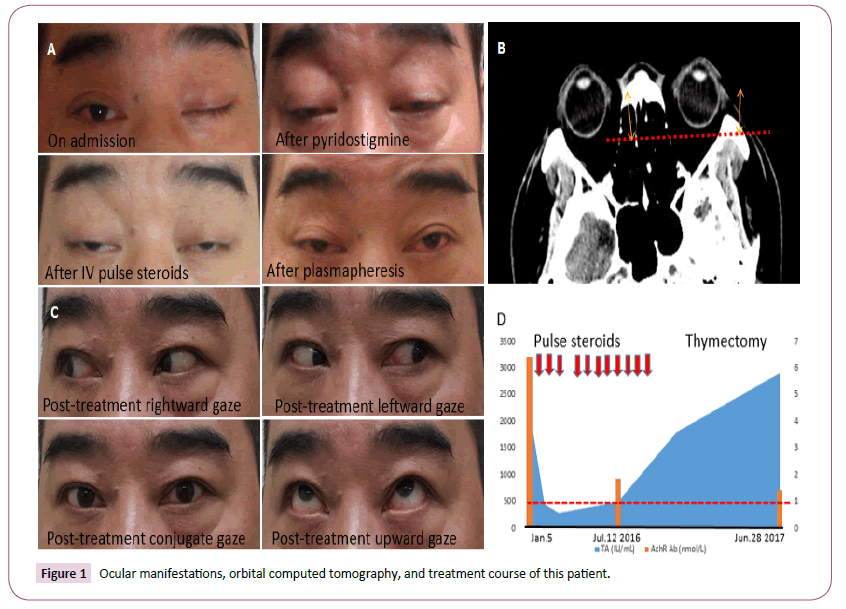
Figure 1: Ocular manifestations, orbital computed tomography, and treatment course of this patient.
The diagnosis of ocular form MG superimposed with autoimmune thyroid disease and associated orbitopathy was made, and he received IV pulse steroid therapy with daily methylprednisolone 500 mg infusion for 3 days, pyridostigmine 60 mg five times a day, oral azathioprine 75 mg once a day (1 mg/kg/day), and pentoxifylline 400 mg twice a day. The ophthalmoplegia and ptosis were stationary (Figure 1A). However, after pulse steroid therapy, he had dyspnea especially when lying down, and the vital capacity progressively declined from 2700 to 1900 ml. Shortness of breath emerged, accompanied with high pCO2 levels, and non-invasive positive pressure ventilation was applied. Double filtration plasmapheresis (DFPP) was started immediately. The vital capacity, ophthalmoplegia and ptosis all improved after four sessions of DFPP (Figure 1A).
His ocular symptoms relapsed occasionally when he felt tired. Otherwise, serum IgG4 level was also elevated (208 mg/dL, reference range <135 mg/dL), but there were no definite masslike lesions detected clinically and in imaging studies. He received eight doses of IV weekly pulse steroid therapy with 250 mg methylprednisolone, and no more deterioration of ocular MG to generalized MG was noted. His ocular symptoms were fully recovered (Figure 1C), accompanied with decreasing levels of AchR Ab but persistent elevation of TA (Figure 1D), indicating that both ocular type of MG and autoimmune thyroid disease contributed to the patient’s ophthalmoplegia and ptosis. However, the association with elevating serum IgG4 and possible IgG4-related disease might suggest a relatively complicated clinical situation of this patient. He underwent right videoassisted thoracoscopic thymectomy on April 22, 2017. Pathology shows mainly adipose tissue, multifocally interspersed with thymic tissue, including lymphoid cells and thymic epithelia, which is consistent with an atrophic thymus. Azathioprine was tapered off, and patient remained symptom-free in November 2017.
Discussion
Acupuncture has been reported to be beneficial in treatment of experimental myasthenia gravis via alteration of acetylcholine receptor expression [3]. However, in our patient, the effect of acupuncture appears to be temporary and limited. Although pulse steroid therapy is effective for the treatment of thyroidassociated ophthalmopathy [4], it is controversial in treating MG. The benefits of pulse steroids have been documented [5], but often require the combination with plasmapheresis and/ or other immunosuppressants [6]. Besides, the possibility of steroid-induced symptom exacerbation, especially deterioration to generalized MG, should be taken into consideration while treating either mild MG or myasthenic crisis [7].
Autoimmune overlaps between MG and other autoimmune disorders reflect common pathogenic mechanisms. One theory is the genetic influence. Early onset MG (<50-year-old) and other autoimmune disorders are associated with the HLA A1-B8-DR3- DQ2 haplotype [8]. An association between the multiple sclerosisassociated allele DRB1*15:01 and the late-onset MG (>50-yearold) has been reported [9]. In our patient, HLA genotyping using reverse sequence-specific oligonucleotide method was performed, and HLA A2/A26, B39/B40, C7/C7, DR8/DR15, and DQ6 were identified. The HLA genotypes did not belong to the high-risk genotype group for early onset MG. Further study is needed to explore the genetic predisposition, if any, for the patient’s complex clinical presentations.
Another proposed theory is that the CD8+ T-cell deficiency underlies the development of chronic autoimmune diseases by impairing CD8+ T-cell control of Epstein-Barr virus (EBV) infection, with the result that EBV-infected autoreactive B cells accumulate in the target organ, where they produce pathogenic autoantibodies and provide costimulatory survival signals to autoreactive T cells which would otherwise die in the target organ by activation-induced apoptosis [10]. Besides, the B lymphotropic virus is a strong polyclonal B-cell activator and may perpetuate the production of autoantibodies that lead to autoimmune diseases [11]. In our patient, the VCA-IgM was negative but the EBNA antibody was positive, indicating that the patient had a past EBV infection [12]. The patient received thymus excision, and there was no evidence of local EBV viral infection in the atrophic thymus. However, we did find the patient to have a relatively low CD8/CD4 ratio of 0.385 (normal 0.5-1).
Initially, IV pulse steroids resulted in marked deterioration to generalized MG in this patient, which was reversed by plasma exchange therapy. Azathioprine has been reported to help prevent the exacerbation of general MG [7]. Unfortunately, the patient’s ocular symptoms recurred under oral immunosuppressants including azathioprine, hydroxychloroquine, and prednisolone. Elevated IgG4 level was also detected, which suggested a more complicated clinical situation that might be responsive to higher dose of steroids [13]. The repeated use of IV pulse steroid treatments did not lead to any recurrence of generalized MG, but resulted in full recovery of the ocular symptoms. In a randomized trial, thymectomy improved clinical outcomes over a 3-year period even in patients with non-thymomatous myasthenia gravis [14], with respect to clinical outcomes, requirements for prednisone and azathioprine, side effects related to immunosuppressive agents, and hospitalization due to disease exacerbations. Though imaging studies did not reveal any thymic pathology, we suggest the patient to undergo thymectomy. After surgical removal of the atrophic thymus, we tapered off azathioprine, and there was no recurrence of myasthenia gravis at least until seven months after the surgery [15].
In Figure 1A Left eyelid total and right eyelid partial ptosis were noted on admission; the extraocular movements were absent and both eyes were fixed at central position. After pyridostigmine, steroid, and plasmapheresis, the ocular symptoms, and signs slightly improved. (B) Brain computed tomography showed mild proptosis of bilateral eyes. The distance from the inter-zygomatic line (dashed line) to the anterior surface of the globe was 22.5 mm on the right eye and 22.1 mm on the left eye (bidirectional arrows); while the distance from the inter-zygomatic line (dashed line) to the posterior sclera was 3 mm (normal 9.9 ± 1.7), which was less than one-seventh of the distance anterior to the inter-zygomatic line (normally more than one-third of the globe was posterior to the inter-zygomatic line) [15]. There was no significant extraocular muscle hypertrophy. (C) The patient’s ocular symptoms and signs fully recovered, with intact rightward, leftward, and upward ocular movements and marked improvement of bilateral eyelid ptosis during outpatient clinic follow-ups. (D) After about six months of steroid treatment, including oral prednisolone and eleven times of intravenous pulse steroid therapy (downward arrows), oral pyridostigmine, azathioprine, and pentoxifylline, and then thymectomy, acetylcholine receptor autoantibodies (AchRAb) declined gradually, but thyroglobulin antibody (TA) levels persistently elevated. Red dotted line indicates normal level of AchRAb.
Conclusion
Myasthenia gravis, which is now considered one of the autoimmune diseases, is highly associated with other autoimmune diseases. Among patients with thyroid associated orbitopathy, ocular form of MG has been linked to autoimmune thyroiditis. Therefore, one diagnosis could be insufficient while making the decision on treatment choices in patients with combined eyelid ptosis, proptosis and ophthalmoplegia. Meanwhile, steroid-induced symptom exacerbation should be taken into consideration while initially treating the relatively elderly patients with severe MG [7]. However, the phenomenon of persistent ocular symptoms disappeared after repeated doses of IV pulse steroid, indicating that early deterioration should not preclude this beneficial treatment option during later followups of these patients [13]. We propose that the IV pulse steroid treatments were even more indicated if an IgG4 disease co-exists with MG and thyroid-associated orbitopathy. Besides, surgical removal of thymoma, normal or even atrophic thymus might be beneficial in patients with high acetylcholine receptor antibodies [14], which is supported by this case experience.
Funding Statement
This study was supported by “Liver Disease Prevention & Treatment Research Foundation, Taiwan” to Jin-Ying Lu.
21161
References
- Berrih-Aknin S (2014) Myasthenia gravis: Paradox versus paradigm in autoimmunity. J Autoimmun 52: 1-28.
- Jokisch F, Kleinlein I, Haller B, Seehaus T, Fuerst H, et al. (2016) A small subgroup of Hashimoto's thyroiditis is associated with IgG4-related disease. Virchows Arch 468: 321-327.
- Huang HP, Pan H, Wang HF (2016) “Warming yang and invigorating qi acupuncture alters acetylcholine receptor expression in the neuromuscular junction of rats with experimental autoimmune myasthenia gravis. Neural Regen Res 11: 465-468.
- Liu X, Wang S, Qin L (2016) Short and long-term effects of high-dose intravenous methylprednisolone pulse therapy on thyroid-associated ophthalmopathy. Exp Ther Med 12: 901-908.
- Kataoka H, Furiya Y, Ueno S (2011) Complete remission of generalized myasthenia gravis by corticosteroid treatment alone without thymectomy. Case Rep Neurol 3: 239-241.
- Nagane Y, Suzuki S, Suzuki N, Utsugisawa K (2011) Early aggressive treatment strategy against myasthenia gravis. Eur Neurol 65: 16-22.
- Melzer N, Ruck T, Fuhr P (2016) Clinical features, pathogenesis, and treatment of myasthenia gravis: A supplement to the guidelines of the german neurological society. J Neurol 263: 1473-1494.
- Nacu A, Andersen JB, Lisnic V, Owe JF, Gilhus NE (2015) Complicating autoimmune diseases in myasthenia gravis: A review. Autoimmunity 48: 362-368.
- Gregersen PK, Kosoy R, Lee AT (2012) Risk for myasthenia gravis maps to a (151) Pro>Ala change in TNIP1 and to human leukocyte antigen-B*08. Ann Neurol 72: 927-935.
- Pender MP (2012) CD8+ T-cell deficiency, Epstein-Barr virus infection, vitamin D deficiency, and steps to autoimmunity: A unifying hypothesis. Autoimmun Dis.
- Ferraccioli G, Tolusso B (2007) Infections, B cell receptor activation and autoimmunity: Different check-point impairments lead to autoimmunity, clonal B cell expansion and fibrosis in different immunological settings. Autoimmun Rev 7: 109-113.
- Linde A, Falk KI (2007) Epstein-Barr virus. In: Manual of Clinical Microbiology. (9th edn). Barron EJ, Jorgensen JH, Landry ML (eds). ASM Press, USA. pp. 1564-1573.
- Glass LR, Freitag SK (2015) Management of orbital IgG4-related disease. Curr Opin Ophthalmol 26: 491-497.
- Wolfe GI, Kaminski HJ, Aban IB (2016) Randomized trial of thymectomy in myasthenia gravis. N Engl J Med 375: 511-522.
- Kralik SF, Kersten R, Glastonbury CM (2012) Evaluation of orbital disorders and cranial nerve innervation of the extraocular muscles. Magn Reson Imaging Clin N Am 20: 413-434.

