Keywords
Posterior cortical atrophy; Benson's syndrome; Lewy bodies disease; Creutzfeldt-Jakob disease
Introduction
Posterior Cortical Atrophy (PCA), also called Benson’s syndrome [1], is a neurodegenerative disease characterized by a progressive occipital and parietal dysfunction, including visuospatial and visuoperceptual disorders, apraxia, alexia, acalculia and language deficit, with relative sparing of memory [2]. Generally PCA is considered as a rare, atypical variant of Alzheimer’s disease, because the most frequent pathologic findings are the betaamyloid neuritic plaques and the tau neurofibrillary tangles; nevertheless other etiologies could be responsible, such as Cortico-Basal Degeneration (CBD), Lewy Bodies Disease (LBD) and Creutzfeldt-Jacob disease (CJD) [3]. Diagnosis remains problematic, and actually it is based on detection of the specific clinical features supported by neuroimaging, which show characteristic posterior brain abnormalities.
Case Report
A 62-year-old right-handed female ex-novo developed a visual disorder, presenting as difficulty reading and spatial disorientation, associated with ideo-motor decline, with slowness in the movements and inconstant confusion. Her remote history was positive for hypertension; familial history was negative for neurodegenerative or psychiatric disease. She was an elementary school teacher with a high educational level (13 years of schooling), and she was temporarily away from work for severe anxious-depressive syndrome derived from her neurological disorder. After an ophthalmologic examination, which ruled out ocular deficit, her General Practioner started an antidepressant therapy with paroxetine at the dose of 20 mg/day, without benefit. Due to the persistence of the disturbances, she quickly had need assistance in daily living activities; for this reason, she was brought to neurological evaluation at our Alzheimer Evaluation Units of Neurological Clinic, approximately six months after the first symptoms. During the first visit the patient arrived in an inadequate hygienic condition and she seems older than her real age. At the first neurological examination she had problems in visual fixation and difficulty to execute simple orders, needing help during the change of position inside the room. She seemed functional blindness: she was unable to reach the objects with the hands, but able to name those objects if presented singularly. These symptoms configured a profile of pure Balint’s syndrome, with optic ataxia (more evident for the left regions), oculomotor apraxia and simultanagnosia. No other neurological signs were evident, in particular strength, sensitivity, coordination, tendon reflexes and cranial nerves were normal. No signs of frontal liberation were observed, in particular no grasping reflex, glabellar and snout reflexes.
The first neuropsychological assessment showed performance below normal limit in Mini Mental State Examination (22/30) with errors in the subtests that evaluate spatial orientation, calculation, reading and writing, constructional praxis; a detailed neuropsychological evaluation confirmed the presence of deficits in perception of objects, constructional apraxia, dysgraphia and alexia (Figure 1); as expected, performances below normal were obtained in tests which evaluate attention, working memory and reasoning and which require the use of visual material (such as Attentive Matrix, Corsi Blocks Tapping Task, Raven’s Progressive Matrices).
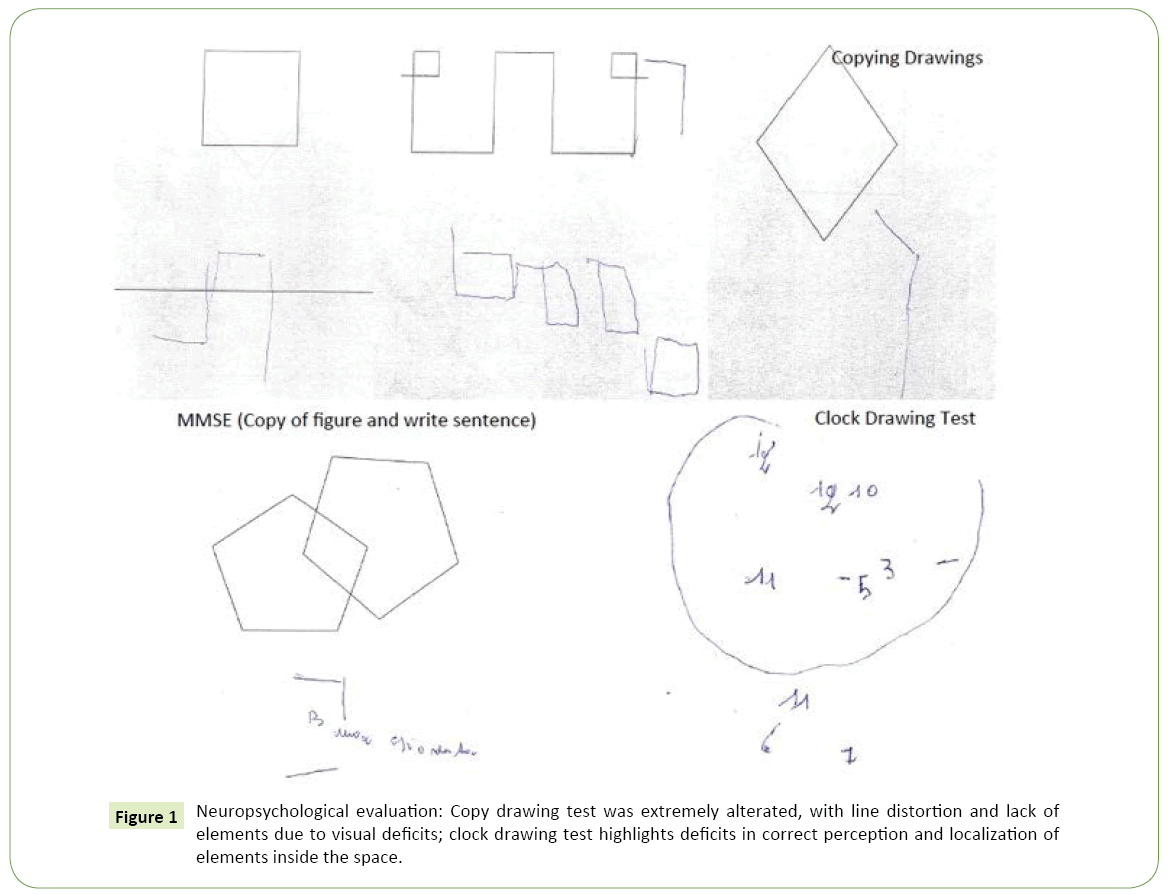
Figure 1: Neuropsychological evaluation: Copy drawing test was extremely alterated, with line distortion and lack of elements due to visual deficits; clock drawing test highlights deficits in correct perception and localization of elements inside the space.
The patient was then subjected to laboratory tests and imaging studies. Laboratory studies were negative for inflammatory or autoimmune diseases, thyroid diseases, metabolic disorders or deficiencies; serology for HIV, Lyme disease and Syphilis, so as cancer markers were also negative. Brain Magnetic Resonance Imaging (MRI) showed predominant parieto-occipital atrophy, particularly evident in the left hemisphere.
Considering the onset with visuospatial and visuoperceptual deficits, the presence of a complex visual disorder, the sparing of memory, the exclusion of other causes, according to criteria identified by Crutch et al. [4], a diagnosis of PCA was supposed.
Eight months later, in consequence of a marked and fast clinical worsening, she was hospitalized in Our Neurological Clinic. Neurological examination was characterized by pyramidal (diffusely brisk reflex) and extrapyramidal (resting and postural bilateral tremor, wrist trochlea with plastic hypertone predominant on right arms) signs, the visual disturbance described above, frontal signs such as grasping reflex and myoclonus at upper limbs. Neuropsychological evaluation showed a moderate cognitive impairment with deficits in all explored domains, including memory, language, attention and executive functions. The patient was subjected to lumbar puncture, which revealed the following data: TAU 420 pg/ml (n.v. <450 pg/ml), pTAU 62 pg/ml (n.v <61 pg/ml), βAmyloid 317 pg/ml (n.v. >500 pg/ml), compatible with an Alzheimer-type pathology; the 14.3.3 protein, tested to exclude prionic disease in consideration of the presence of myoclonus and the rapid worsening of neurological deficits, was negative; other CSF parameters (cell number, proteins level, glucose, pressure) were normal. A second MRI showed more pronounced parieto-occipital atrophy, always more in the left hemisphere (Figure 2). FDG-PET showed a marked posterior hypo-metabolism, bilaterally (Figure 3). The EEG showed diffuse slowing in theta rhythm, without epileptic discharges. So, the diagnosis of PCA was confirmed. The patient was subject to genetic investigations: genetic mutations for MAPT, c9Orf72 and PGRN were negative. Myoclonus was well treated with levetiracetam 500 mg/twice a day, with a good response. For the neurocognitive impairment we started donepezil 10 mg/day, well tolerated by the patient. She is on trimestral follow up at our Clinic since two years: progressively, the neurologic state stabilized in its severity. At the last evaluation the patient’s neurological exams showed an asymmetric rigid-acinetic extrapyramidal syndrome, predominant on right, with well-known visuo-spatial deficits, pyramidal and frontal signs, and she is actually totally depending on caregiver in daily living activities.
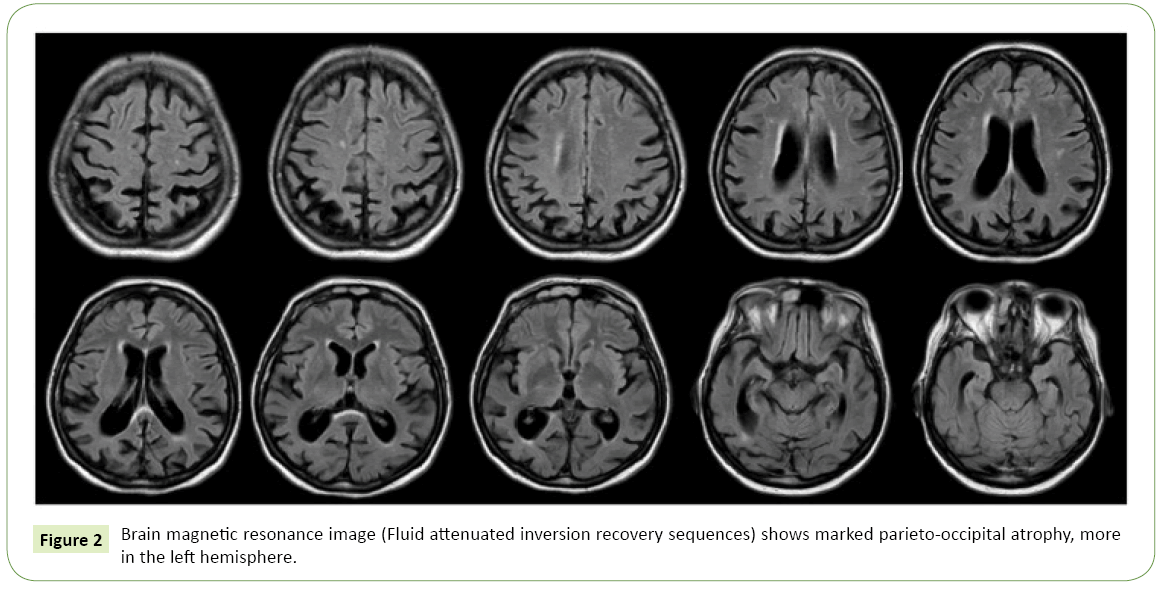
Figure 2: Brain magnetic resonance image (Fluid attenuated inversion recovery sequences) shows marked parieto-occipital atrophy, more in the left hemisphere.
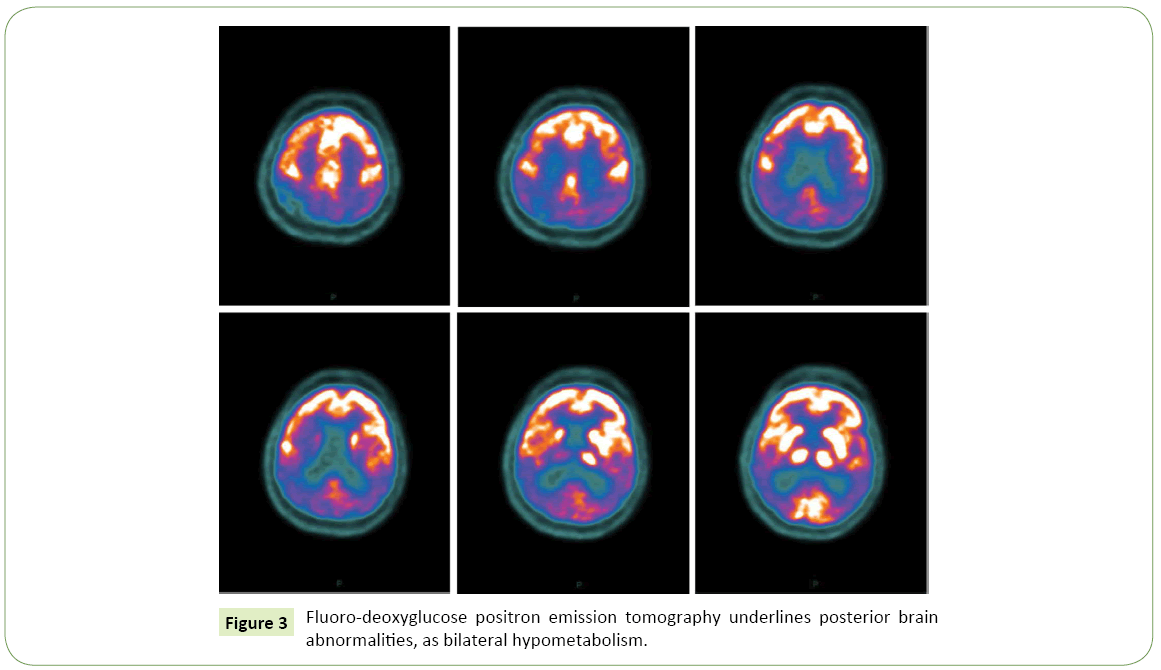
Figure 3: Fluoro-deoxyglucose positron emission tomography underlines posterior brain abnormalities, as bilateral hypometabolism.
Discussion
Posterior Cortical Atrophy (PCA) is a neurodegenerative syndrome characterized by progressive dysfunction of posterior cortical regions. It is usually considered the posterior variant of Alzheimer’s Disease, which often shares the same pathogenesis; generally the patients with PCA are younger than AD patients (peak of incidence: 50-60 years) with a better prognosis and a slower course [5,6]. PCA is clinically characterized by a progressive impairment of visuo-spatial and visuo-perceptual functions in the absence of ocular deficits. These signs, which characterized the disease at the onset, are frequent associated with mood disorders and impairment of daily living activities and misdiagnosed and confused with oculistic, psychiatric or geriatric disturbances [7,8]. Other common signs are linked to dysfunction of the posterior areas of the brain: simultagnosia, alexia, dysgraphia, difficulties in spatial orientation and driving; Balint’s and Gerstmann’s syndromes are frequent [4]. At the onset memory and executive functions are usually spared; commonly the neurological examination is unremarkable, but some patients can develop sensorimotor signs, which remain less frequent, except in other etiology than AD, as CBD or LBD [9]; myoclonus, rigidity and tremor are rare [9].
In PCA neuroimaging generally show characteristic posterior brain atrophy, including occipital, parietal and posterior temporal lobes; the right hemisphere is more commonly affected than the left one [10]. Functional studies help in diagnosis, showing bilateral parietal and occipital hypo-metabolism [11].
Neuropsychological evaluation is necessary to early identify the visuospatial and praxis disorders; many patients show difficulties in objects perception and in space analysis, which are evident in tests of copying drawing [12]. Moreover, at the onset difficulties in writing and reading (dyslexia, dysgraphia and alexia) are predominant, possibly associated with dyscalculia and limb apraxia; the develop of deficits in memory, language and executive functions are usually late.
CSF analysis shows, in most cases, low level of beta-amyloid and increased level of TAU, confirming a diagnosis of amyloid-based dementia as an atypical AD [13].
PCA is reported as sporadic disorder but the genetic research could be useful in the atypical cases, to confirm the clinical hypothesis [14,15]. A recent classification distinguishes two variants of PCA: a pure-PCA, which meets the clinic-radiological criteria for PCA; a PCA-plus, with additional features that direct diagnosis to CBD, LBD or CJD [16].
Conclusion
In our case clinical onset with complex visual symptoms, and neuroimaging with posterior brain atrophy, oriented to a case of pure-PCA. Subsequently the development of uncommon symptoms, such as extrapyramidal signs and primitive reflexes, has complicated the clinical profile, requiring more diagnostic workup with neuropathologic, genetic and neuroradiologic detailed analysis, to confirm an atypical Alzheimer-like dementia. Aetiologic diagnosis remains important for a prognostic point of view, above all in case of rapidly worsening dementia with infrequent signs, to exclude overlap syndrome.
19537
References
- Benson DF, Davis RJ, Snyder BD (1998) Posterior cortical atrophy. Arch Neurol 45: 789-793.
- Tang-Wai DF, Graff-Radford NR, Boeve BF, Dickson DW, Parisi JE, et al. (2004) Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology63: 1168-1174.
- Renner JA, Burns JM, Hou CE, McKeel DW Jr, Storandt M, et al. (2004) Progressive posterior cortical dysfunction: Aclinico-pathologic series. Neurology63: 1175-1180.
- Crutch SJ, Schott JM, Rabinovici GD, Boeve BF, Cappa SF, et al. (2013) Shining a light on posterior cortical atrophy. Alzheimers Dement9: 463-465.
- Slattery CF, Crutch SJ, Schott JM (2015)Phenotypical variation in Alzheimer's disease: Insights from posterior cortical atrophy. PractNeurol15: 2-4.
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, et al. (2012) Posterior cortical atrophy. Lancet Neurol11: 170-178.
- Ishiwata A, Kimura K (2016) Homonymous hemianopsia associated with probable Alzheimer's disease. J Nippon Med Sch83: 87-92.
- Everhart DE, Highsmith JM, Davis CE (2012) Posterior cortical atrophy: A case study of Benson's syndrome that initially presented as anxiety disorder. ApplNeuropsychol Adult19: 229-236.
- Ryan NS, Shakespeare TJ, Lehmann M, Keihaninejad S, Nicholas JM, et al. (2014) Motor features in posterior cortical atrophy and their imaging correlates. Neurobiol Aging35: 2845-2857.
- Millington RS, James-Galton M, Maia Da Silva MN, Plant GT, Bridge H (2017) Lateralized occipital degeneration in posterior cortical atrophy predicts visual field deficits. NeuroimageClin14: 242-249.
- Singh TD, Josephs KA, Machulda MM, Drubach DA, Apostolova LG, et al. (2015) Clinical, FDG and amyloid PET imaging in posterior cortical atrophy. J Neurol262: 1483-1492.
- Boban M, Mioc M (2014) Upside-down drawing in posterior cortical atrophy. Can J NeurolSci41: 654-665.
- Arighi A, Rango M, Bozzali M, Pietroboni AM, Fumagalli G, et al. (2015) Usefulness of multi-parametric MRI for the investigation of posterior cortical atrophy. PLoS One10: e0140639.
- Caroppo P, Belin C, Grabli D, Maillet D, De Septenville A, et al. (2015) Posterior cortical atrophy as an extreme phenotype of GRN mutations. JAMA Neurol72: 224-228.
- Peng G, Liu P, He F, Luo B (2016) Posterior cortical atrophy as a primary clinical phenotype of corticobasal syndrome with a progranulin gene rs5848 TT genotype. Orphanet J Rare Dis11: 13.
- Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, et al. (2017) Consensus classification of posterior cortical atrophy. Alzheimers Dement.

