Keywords
Visceral leishmaniasis: PLGA-Nanoparticles; Amphotericin B; Doxorubicin; Lectin Introduction
Introduction
PLGA-Nanoparticle (PLGA-NPs) based combination therapy has the potential to overcome the toxicity and poorly controlled dosing of traditional systemic combination therapies against visceral leishmaniasis [1]. Though NP based therapeutics for visceral leishmaniasis (VL) therapy have been the subject of many investigations over the past several years, ratio metric delivery and harmonized release of multiple drugs from single PLGA-NPs scaffolds remain difficult challenges. Many of the most studied NPs manners for delivery e.g., liposomes, dendrimers and micelles are not readily open to incorporation and release of multiple drugs. Due to the complex interactions between drugs in living systems, a NPs platform for specific tuning and rapid dissimilarity of drug loading ratios and release kinetics would enable the discovery of optimal formulations for specific VL types [2]. We view this challenge as an artificial problem: multidrug-loaded NP synthesis would be most ecient if serial particle conjugation and encapsulation reactions were replaced with highly convergent approaches wherein the main elements of a desired NP (e.g., drug molecules) are used to figure particles directly. Herein we present a unique strategy that uses carefully designed drug as building blocks for the macrophage targeted multi-drug-loaded NPs against VL.
Our PLGA-NPs bring precise ratios of Amphotericin B (AmB), and Doxorubicin (DOX). These drugs were chosen due to their non-coinciding toxicity profiles. The most severe dose-limiting side effects from doxorubicin arise from cardio toxicity, while those from amphotericin B result from neurotoxicity and myelo suppression respectively [2,3]. Thus, maximum therapeutic index could be attained, in principle, via simultaneous dosing of each drug at or near its maximum tolerated dose (MTD). We show that two-drug-loaded PLGA-NPs with ratios matched to multiples of the MTD of each drug overtake analogous one and two-drug-loaded PLGA-NPs in invitro studies. Our synthesis relies on the double emulsion method, which permits the preparation of two drugs with different solubility characteristics.
Nanoparticle formulations in which drugs themselves aid as carriers provide a number of interesting opportunities; when compounded through double emulsion method, insoluble drugs can form homogeneous colloids with tunable sizes and shapes that simply are not possible with standard FDA approved carriers.
Materials and Methods
Drugs and chemicals
AmB was obtained as a gift sample from M/s Ambalal Sarabhai Enterprises, Vadodara, India. Doxorubicin was received as gift sample from Sun Pharmaceuticals Industries Limited, India. PLGA 50:50 (intrinsic viscosity 0.35 dl/g) was purchased from Boehringer Ingelheim (Ingelheim, Germany). Span 85, Tween 20, sodium chloride, potassium dihydrogen phosphate, Polyvinyl alcohol (PVA), DMSO, Isopropyl alcohol, Eagle’s medium, Locke’s solution and dialysis bag membrane (MWCO: 3500) were obtained from Sigma Chemicals, USA. Chloroform and all other chemicals were of pure analytical grade and used as procured.
Preparation method of PLGA-NPs
To explore the possibility of this approach, we designed and synthesized PLGA-NPs. With this pool of novel drugs in hand, we targeted macrophages with each of drug that correspond to 2 times the individual drugs. PLGA nanoparticles were formulated according to double emulsion method with slight modification as per laboratory setup [4]. In brief, In a plastic vial Amphotericin B was dissolved in DMSO and in a separate vial doxorubicin was dissolved in water to form the aqueous phase, which was then added to a solution of PLGA in DMSO to give a w/o emulsion which was then sonicated and added drop wise under stirring to aqueous solution containing 0.2% PVA to form the secondary emulsion. The secondary emulsion was again sonicated to reduce the particle size and then was diluted with sufficient water to help solvent diffusion and precipitation of the polymer resulting into formation of NPs. The resulting nanoparticle suspension was used immediately for analysis or lyophilized for storage at 4°C.
PLGA-NPs were optimized for various parameters. These include the drug content (AmB and DOX), polymer content (PLGA), and sonication time. At higher concentrations, (10-20 mg of AmB and 15-20 mg of DOX) particles do not formed within Nano-size range. However, as the concentration of AmB and DOX was gradually lowered, relative numbers of particle size decreased and nanoparticles were formed (Tables 1a and 1b). Similarly, as the concentration of AmB and DOX was gradually lowered, particle size was decreased while percent drug entrapment was increased. When concentration of AmB and DOX was used at 6 and 10 mg respectively, PLGA-NPs formulations were free of other undesired structures, average particle size measured was 356.2 ± 0.04 nm and entrapment efficiency was recorded to be 78.4 ± 2.01 for AmB and 70.06 ± 0.14 for DOX respectively. On the basis of minimum particle size and maximum percent drug entrapment formulations PLGA-NP4 was considered to be optimum. Formulations with optimum AmB and DOX content were subjected to sonication for different time periods to optimize the sonication time. Figure 1 shows that as the sonication time was increased from 0 to 15 min, average particle size was recorded to be decreased. From this hypothesis, optimum sonication time was recorded to be 15 min, which gave particle size of 356.2 ± 0.04 nm. On further increasing the sonication time (i.e., at 15 min) beyond the optimum limit the particle size was recorded to be 134.8 ± 0.02 nm and PLGA-NPs might have localized in hepatocytes apart of macrophages, the target site. Formulation having optimized AmB and DOX, sonication time were subjected to optimization of polymer content (PLGA). Figures 2a and 2b shows that with an increase in PLGA content in PLGA-NPs, distinctive percent entrapment of AmB and DOX were recorded. As the PLGA content was increased from 50-200 mg drug entrapment was also recorded to be increased. However, it may be attributed to the subsequent decreased stability of the PLGA-NPs with the increase in PLGA content and initial fast release of drug from these unstable particles [4]. Optimum PLGA content was found to be 50 mg which could entrap maximum amount of drug AmB and DOX respectively.
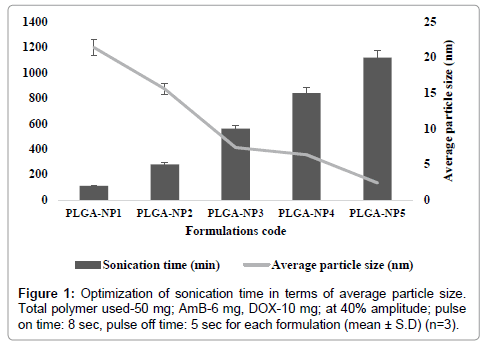
Figure 1: Optimization of sonication time in terms of average particle size. Total polymer used-50 mg; AmB-6 mg, DOX-10 mg; at 40% amplitude; pulse on time: 8 sec, pulse off time: 5 sec for each formulation (mean ± S.D) (n=3).
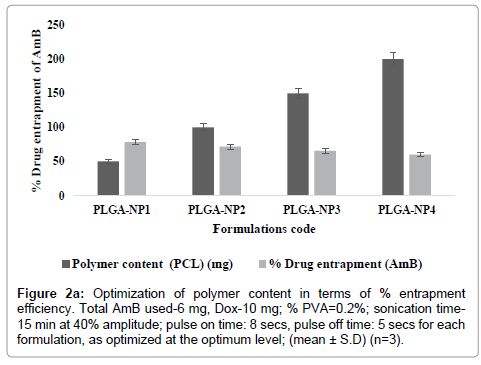
Figure 2a: Optimization of polymer content in terms of % entrapment efficiency. Total AmB used-6 mg, Dox-10 mg; % PVA=0.2%; sonication time- 15 min at 40% amplitude; pulse on time: 8 secs, pulse off time: 5 secs for each formulation, as optimized at the optimum level; (mean ± S.D) (n=3).
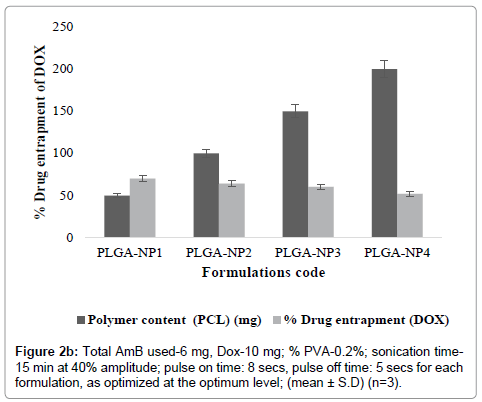
Figure 2b:Total AmB used-6 mg, Dox-10 mg; % PVA-0.2%; sonication time- 15 min at 40% amplitude; pulse on time: 8 secs, pulse off time: 5 secs for each formulation, as optimized at the optimum level; (mean ± S.D) (n=3).
PLGA-NPs were optimized for drug content, polymer content, sonication time. AmB and DOX to polymer content was optimized by keeping PLGA content 50 mg, and sonication time at constant levels while AmB and DOX content was varied at different weight levels, i.e., (2, 4, 6, 8 and 10 mg) whereas for DOX (2, 5, 10, 15 and 20 mg) respectively in different formulations for determining optimum AmB and DOX content [5]. Average particle size of different formulations was measured by photon correlation spectroscopy using Zetasizer Nanoseries (Nano-ZS 90, Malvern Inst. Ltd. UK) using a flow-through cell and percent drug entrapment in different formulations was also determined [6]. PLGA-NPs with optimum AmB and DOX content were optimized for optimum polymer content (PLGA) in terms of percent drug entrapment. AmB (6 mg) and DOX content (10 mg), sonication time (12 min) were kept constant while polymer (PLGA) content was varied for different formulations. PLGA-NPs with optimum AmB and DOX content and polymer content (PLGA) were optimized for optimum sonication time in terms of average particle size. PLGA content, and both drugs (AmB and DOX) were kept constant while sonication time was varied (i.e., 0, 2, 4, 6, 8, 10, 12 and 15 min) for different formulations. PLGA-NPs were evaluated for average particle size as described above. Drug content, polymer content and sonication time, however, were kept constant at its optimum level. PLGA-NPs were evaluated for percent drug entrapment and percent drug release as reported [7].
Coating with lectin
To conjugate the surface of PLGA-NPs with lectin modified emulsion- solvent method was performed [8]. Lectin incorporated PLGA-NPs was prepared by incubating PGNP-NPs with 1 ml lectin solution in 10 mM HEPES buffer (pH 7.4) at 37°C for 24 hrs under sonication. The unattached lectin was removed by washing followed by centrifugation of the nanoparticle suspension; the whole process being repeated thrice to ensure complete removal. The Lectin concentration was optimized by measuring the change in zeta potential of the lectin coated PLGA-NPs dispersion in deionized water at 25°C [9].
In-vitro characterization of PLGA-NPs
Average particle size and size distribution of PLGA-NPs and Lectin- PLGA-NPs were analyzed by photon correlation spectroscopy (PCS) at 25°C under an angle of 90°. Zeta potential was determined using Laser Doppler anemometry (LDA) at 25°C. The PCS and LDA analysis were performed using a Zetasizer NanoZS (Malvern Instrument, Worcestershire, UK), after dispersing in deionized water [10].
In order to quantify the percent drug entrapment encapsulated amount, AmB and DOX was extracted from the PLGA-NPs, diluted with methanol and analyzed using HPLC. The HPLC (LC-10ATvp, Shimadzu, Tokyo, Japan) was equipped with a Lichrosphere reverse-phase C18 column (250×4 mm, 5 μm; Merck, Darmstadt, Germany). Acetonitrile with KH2PO4 buffer (pH 3.5, adjusted with orthophosphoric acid), (60:40, v/v) was employed as mobile phase at 1.0 ml/min flow rate and column effluent was detected with a UV detector at 405 and 235 nm respectively. Results are expressed as AmB and DOX actual loading (drug amount encapsulated per 100 mg of PLGA-NPs) and encapsulation efficiency (EE) [11].
The in-vitro AmB and DOX release was performed using dialysis membrane diffusion technique. Briefly, AmB and DOX equivalents of total amount of drug encapsulated formulation was suspended in 1 ml of phosphate buffer solution (PBS, pH 7.4) in a dialysis bag and dialyzed against 250 ml PBS with 0.5% Tween 80 contained in dissolution apparatus (DISSO 2000, Labindia, India), thermostated at 37 ± 1 ºC with moderate shaking at 100 rpm. At specific time intervals, a definite volume (1 ml) of the release medium was withdrawn and replenished with fresh PBS and analyzed for AmB and DOX amount using validated HPLC method. Each measurement was performed in triplicate and reported as their average [12].
Stability study
The stability studies were performed by observing drug leaching and change in particle size following incubation of PLGA-NPs with freshly pooled rat serum at 37 ± 1°C. The drug content of the PLGANPs was determined by the method described previously with slight modifications. PLGA-NPs formulations were incubated with 2 ml serum at 37 ± 1°C for 1, 2, 4, 6 and 24 h. After specified time intervals, suspensions were centrifuged at 20,000 rpm for 15 min and supernatant was filtered through 0.22 μm membrane filter. The filtrate was analyzed for drug content by reverse phase high performance liquid chromatography method as described elsewhere [5,7,11]. The particle size of PLGA-NPs formulations was determined after 24 h incubation of the formulations with the serum using particle size analyzer.
Results and Discussion
Preparation and in-vitro characterization of PLGA-NPs
PLGA-NPs formulations, having combination of dual agents (AmB and DOX) were prepared by modified double emulsion method with slight modification as per our laboratory set up [9]. These include the drug content (AmB and DOX), polymer content (PLGA), and sonication time. At higher concentrations, particles do not formed within size range. However, as the concentration of AmB and DOX was gradually lowered, relative numbers of particle size decreased and nanoparticles were formed (Tables 1a and 1b). Similarly, as the concentration of AmB and DOX was gradually lowered, particle size was decreased while percent drug entrapment was increased. When concentration of AmB and DOX was used at 6 and 10 mg respectively, PLGA-NPs formulations were free of other undesired structures, average particle size measured was 356.2 ± 0.04 nm and entrapment efficiency was recorded to be 78.4 ± 2.01 for AmB and 70.06 ± 0.14 for DOX respectively. On the basis of minimum particle size and maximum percent drug entrapment formulations PLGA-NP4 was considered to be optimum. Formulations with optimum AmB and DOX content were subjected to sonication for different time periods to optimize the sonication time. Figure 1 shows that as the sonication time was increased from 0 to 15 min, average particle size was recorded to be decreased. From this hypothesis, optimum sonication time was recorded to be 15 min, which gave particle size of 356.2 ± 0.04 nm. On further increasing the sonication time (i.e., at 15 min) beyond the optimum limit the particle size was recorded to be 134.8 ± 0.02 nm and PLGA-NPs might have localized in hepatocytes apart of macrophages, the target site. Formulation having optimized AmB and DOX, sonication time were subjected to optimization of polymer content (PLGA). Figures 2a and 2b shows that with an increase in PLGA content in PLGA-NPs, distinctive percent entrapment of AmB and DOX were recorded. As the PLGA content was increased from 50-200 mg drug entrapment was also recorded to be increased (Figures 2a and 2b). However, it may be attributed to the subsequent decreased stability of the PLGA-NPs with the increase in PLGA content and initial fast release of drug from these unstable particles [2,11]. Optimum PLGA content was found to be 50 mg which could entrap maximum amount of drug 78.4 ± 2.01 for AmB and 70.06 ± 0.14 for DOX respectively.
| Formulation code |
AmB content (mg) |
% Entrapment efficiency of AmB |
Avg. particle size (nm) |
| PLGA-NP1 |
2 |
65.4±1.08 |
948.8±0.06 |
| PLGA-NP2 |
4 |
70.2±1.62 |
781.2±0.03 |
| PLGA-NP3 |
6 |
78.4±2.01 |
340.6±0.08 |
| PLGA-NP4 |
8 |
80.1±2.04 |
280.7±0.02 |
| PLGA-NP5 |
10 |
81.01±1.82 |
206.1±0.08 |
Table 1a: Optimization of Amphotericin B(AmB).Total polymer used-50 mg, DOX-10 mg, % PVA- 0. 2 (50 ml), sonication time-15 min; at 20% amplitude; pulse on time: 5 min, pulse off time: 5 sec for each formulation (mean±S.D) (n=3).
| Formulation code |
DOX content (mg) |
% Entrapment efficiency (DOX) |
Avg. particle size (nm) |
| PLGA-NP1 |
2 |
50.4±0.02 |
948.8±0.06 |
| PLGA-NP2 |
5 |
61.8±0.10 |
781.2±0.03 |
| PLGA-NP3 |
10 |
70.06±0.14 |
340.6±0.08 |
| PLGA-NP4 |
15 |
72.01±0.10 |
280.7±0.02 |
| PLGA-NP5 |
20 |
78.03±0.12 |
206.1±0.08 |
Table 1b: Optimization of Doxorubicin (DOX).Total polymer used-50 mg, AmB- 6 mg, % PVA- 0.2 (50 ml), sonication time-15 min; at 20% amplitude; pulse on time: 5 min, pulse off time: 5 secs for each formulation (mean±S.D) (n=3).
After optimizing the process parameters, PLGA-NPs were coated with macrophage specific ligand Lectin. Moreover, Lectin possessed a positive charge, while PLGA-NPs were negatively charged which further facilitated the adsorption process and resulted in a reduction of the zeta potential of the dispersion. For PLGA-NPs the initial positive value of the zeta potential was decreased on addition of cationic ligand lectin and approached towards a minimum value of 8.44 mV at 03:02 lectin: polymer (w/w) ratio (Figure 3). It was apparently related to the extent of the covering of the surface charge by the lectin. On further addition of lectin especially beyond this optimum ratio no significant change in the zeta potential was occurred for PLGA-NPs. Hence optimum ratio of lectin: polymer was recorded to be 3:2 (w/w) for PLGA-NPs. This indicated that at an optimal 3:2 w/w ratio of lectin: polymer, the integration of lectin was occurred at saturation level. For optimization of incubation time, the PLGA-NPs formulations using the optimum lectin: polymer ratio were incubated with lectin for different time periods (0, 1, 2, 3, 4 and 6, 7, 8 h) and the change in zeta potential was recorded (Figure 4). Zeta potential value declined steeply from its initial value of 20.36 mV for PLGA-NPs, which might have been attributed to the charge quenching of the surface associated with free lectin. With a longer incubation time the amount of residual free lectin was decreased and the change in zeta potential was not significant. This indicated that at the end of 7 hrs the interaction of added lectin could have completed. Table 2 shows the average particle size and percent drug entrapment of optimized formulation of PLGA-NPs and lectin-PLGANPs. The increase in average particle size in case of formulation lectin- PLGA-NPs as compared to formulation PLGA-NPs is an indication of coating, which can be distinguished by dark black boundary of the formulation lectin-PLGA-NPs. Percent drug entrapment of optimized PLGA-NPs and lectin-PLGA-NPs formulation was recorded to be 75.8 ± 1.21 (AmB) and 71.20 ± 0.04 (DOX) and 72.4 ± 2.01 (AmB), 68.04 ± 0.12 (DOX) respectively, revealing that lectin anchoring did not result in significant lowering of the percent drug entrapment. Preformed PLGA-NPs were used for anchoring of ligand and this may presumably be the reason for the insignificant change recorded in the percent drug entrapment value. Relatively high entrapment of AmB and DOX in the PLGA-NPs could be attributed to the lipophilic nature of the drug, since the entrapment was dependent upon lipid: aqueous phase ratio.
| S No |
Parameters |
AmB and DOX loaded PLGA-NPs |
Lectin-PLGA-NPs |
| 1 |
Size(nm) |
340.6 ±0.08 nm |
402.2±0.06 |
| 2 |
Polydispersity index |
0.160 |
0.410 |
| 3 |
Zeta potential (mV) |
-4.82 |
4.82 mV |
| 4 |
% Drug entrapment efficiency |
75.8±1.21 (AmB) and 71.20±0.04 (DOX) |
72.4±2.01 (AmB),
68.04±0.12 (DOX) |
| 5 |
per cent drug release |
AmB release over 12 days DOX release for as long as 8 days. |
AmB release over 10 days DOX release for as long as 6 days. |
(mean±SD) (n=3)
Table 2: Characterization of optimized plain and ligand anchored formulations.
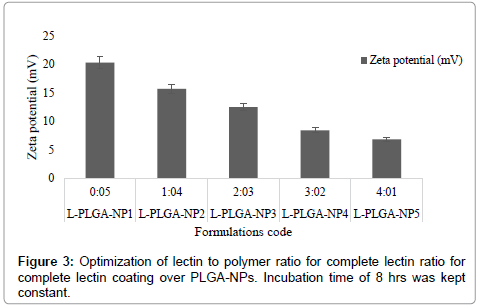
Figure 3: Optimization of lectin to polymer ratio for complete lectin ratio for complete lectin coating over PLGA-NPs. Incubation time of 8 hrs was kept constant.
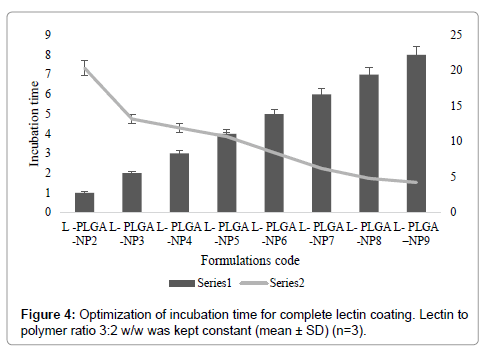
Figure 4: Optimization of incubation time for complete lectin coating. Lectin to polymer ratio 3:2 w/w was kept constant (mean ± SD) (n=3).
Stability studies
Stability of developed PLGA-NPs in serum was measured as percent drug leaching from PLGA-NPs and change in particle size of PLGA-NPs after incubation with serum at 4 ± 1°C, 28 ± 1°C, 37 ± 1°C. The PLGA-NPs were found to be almost stable upon incubation with freshly pooled rat serum. Only 95.2 ± 1.4, 91.2 ± 2.2, 92.6 ± 2.2 and 96.1 ± 2.4, 94.8 ± 2.0, 94.0 ± 2.0 drug was leached into serum after 30 days of incubation at 4 ± 1°C, 28 ± 1°C, 37 ± 1°C from formulation PLGA-NPs and lectin-PLGA-NPs respectively (Figure 5). This may be attributed to the lipophilic nature of the prepared formulation hence prevented the drug leaching in serum [13]. Similarly, particle size analysis of PLGA-NPs did not show any significant change in the particle size of formulation PLGA-NPs and lectin-PLGA-NPs respectively after 30 days incubation with serum. There was only slight increase in the particle size of formulation PLGA-NPs and lectin-PLGA-NPs. The PLGA-NPs formulations developed for targeting to macrophages should be cleared from the circulation within a very short span of time. Therefore, this insignificant increase in particle size in vitro may not have discernible bearing on bio disposition [14,15].
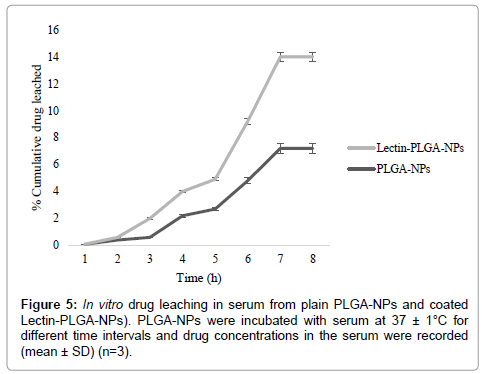
Figure 6: In vitro drug leaching in serum from plain PLGA-NPs and coated Lectin-PLGA-NPs). PLGA-NPs were incubated with serum at 37 ± 1°C for different time intervals and drug concentrations in the serum were recorded (mean ± SD) (n=3).
In this case, the nanoparticles target at the target sites i.e., liver, spleen kidney more efficiently and specifically after incorporation of ligand-lectin. This novel concept for combination delivery is only made possible using highly convergent NP synthesis. This approach has no fundamental limitation in terms of the number and ratio of molecular species that could be built into particles. Through the combination of two drugs in a PLGA-nanoparticle incorporated with ligand lectin to target specifically against target sites of visceral leishmaniasis, libraries of multi-drug-loaded PLGA-NPs can be readily synthesized in parallel for efficacy optimization.
Conclusion
In this paper, we report for the first time that two drugs with different properties can be simultaneously entrapped into PLGANanoparticles with a relatively high-entrapment efficiency and small size. The influences of various processing variables on zeta potential, particle size, drug loading and encapsulation capacity were systematically assessed. Using PLGA- Nanoparticles as a novel drug delivery platform, various strategies for administering AmB/DOX combinations were systematically compared. It was found that the dualagent loaded AmB-DOX- PLGA- Nanoparticles system resulted in efficient macrophage targeted to co-administration of two single-agent loaded PLGA- Nanoparticles (AmB-PLGA-NPs+DOX- PLGA-NPs), than that of the free AmB/DOX-PLGA-NPs and one free drug/another agent loaded PLGA -NPs combination. In conclusion, the proposed PLGA-NPs with combination of dual drugs i.e., AmB and DOX systems show absurd potential for intracellular macrophage targeting. The formulations could considerably alter the pharmacokinetics of AmB and DOX, providing prolonged action at comparatively low drug doses thereby falling the toxicity problems like nephrotoxicity, cardiac arrhythmia etc. The developed systems (plain and Lectin coated PLGA-NPs) appear promising for the treatment of VL specifically. In summary, our findings indicated that Lectin-PLGA-NPs deliver advanced amount of the drug to the desired organ sites due to being an efficient macrophage targeted drug delivery system. Targeted delivery directly reduces the drug dose, which is highly desirable for optimized therapeutic effect and diminished undesirable toxicity.
Acknowledgements
Author Prachi Sharma is thankful to Apeejay Stya University, New Delhi, India, for providing necessary facilities for research work. The help and facilities provided by the Central Drug Research Institute, Lucknow (UP), India are also duly acknowledged.
Declaration of Interest
The authors declare no competing Ânancial interest.
18667
References
- Kumar N, Sharma P, Jaiswal A, Dube A (2016) Development and evaluation of p-aminophenylmannopyranoside anchored emulsomes for treatment of experimental visceral leishmaniasis. Ann ClinCytolPathol 2: 1042.
- Gupta S, Vyas SP (2007) Development and characterization of amphotericin B bearing emulsomes for passive and active macrophage targeting. J Drug Target15: 206-217.
- Mukherjee S, Das L, Kole, L, Karmakar S, Datta N, et al. (2004) Targeting of parasite-specific immunoliposome-encapsulated doxorubicin in the treatment of experimental visceral leishmaniasis. J Infect Dis 189: 1024-1034.
- Khare P, Rastogi P, Gupta S, Maurya R (2014) In vitro and in vivo efficacy of a new herbaceous indian plant-Abutilon indicum against Leishmaniadonovani infection.Amer J PhytomedClinTher 2: 134-139.
- Kumar P, Kumar A, Verma SS, Dwivedi N (2008) Leishmaniadonovanipteridine reductase 1: Biochemical properties and structure-modeling studies. ExpParasitol 120: 73-79.
- Vyas SP, Quraishi S, Gupta S, Jaganathan KS(2005) Aerosolized liposome-based delivery of amphotericin B to alveolar macrophages. Int J Pharm296: 12-25.
- Gupta S, Dube A, Vyas SP (2013) Development and characterization of amphotericin B loaded solid lipid nanoparticles against experimental visceral leishmaniasis.Pharmaceutical Nanotechnology 1: 54-67.
- Yin Y, Chen D, Qiao M, Lu Z, Hu H (2006) Preparation and evaluation of lectin-conjugated PLGA nanoparticles for oral delivery of thymopentin. Journal of Controlled Release 116: 337-345.
- Pal A, Gupta S, Jaiswal A, Dube A (2012) Development and evaluation of tripalmitinemulsomes for the treatment of experimental visceral leishmaniasis. J Liposome Res 22: 62-71.
- Kunjachan S, Gupta S, Dwivedi A, Dube A, Chourasia M (2011) Chitosan-based macrophage-mediated drug targeting for the treatment of experimental visceral leishmaniasis.J microencapsulation 28: 301-10.
- Mukherjee S, Das L, Kole L, Karmakar S, Datta N, et al. (2004) Targeting of parasite-specific immunoliposome-encapsulated doxorubicin in the treatment of experimental visceral leishmaniasis. J Infect Dis 189: 1024-1034.
- Italia JL,Datta P, Ankola DD, Kumar MNVR (2008) Nanoparticles enhance peroral bioavailability of poorly available molecules: epigallocatechin gallate nanoparticles ameliorates cyclosporine induced nephrotoxicity in rats at three times lower dose than oral solution. J Biomed Nanotech 4: 304-312.
- Sharma S, Kumar P, Jaiswal A, Dube A (2011) Development and characterization of doxorubicin loaded microparticles against experimental visceral leishmaniasis. J Biomed Nanotech 7: 135-136.
- Gupta S, Dube A, Vyas SP (2007) Antileishmanial efficacy of amphotericin B bearing emulsomes against experimental visceral leishmaniasis.J Drug Target 15: 437-444.
- Sanchez-Brunete JA, Dea MA, Rama S, Bolas F, Alunda JM (2004) Treatment of experimental visceral leishmaniasis with amphotericin B in stable albumin microspheres. Antimicrob Agents Chemother 48: 3246-3252.


