Beth Harknes* and Hani Fanous
Children's National Health System, Washington DC, USA
*Corresponding Author:
Beth Harkness
Children's National Health System
Washington DC, USA
Tel: 17038671969
E-mail: bharknes@childrensnational.org
Received date: May 25, 2017; Accepted date: June 01, 2017; Published date: June 08, 2017
Citation: Harkness B, Fanous H. A Perspective on Diverse Phenotypic Expression of Two Siblings Affected by Cystic Fibrosis with Homozygous F508del Genotype. Transl Biomed. 2017, 8:2. doi:10.21767/2172-0479.100113
Keywords
F508del; Translational; Diversity; Cystic fibrosis; Newborn screening; Genetics; Genomics; Body mass index; MRSA; FEV1, Growth hormone; Cystic fibrosis; NBS; Phenotype; Variability; Sibling; Study; Deltaf508
Introduction
Cystic Fibrosis (CF) is a single gene recessive genetic disorder caused by mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and has been estimated to have a prevalence of 1/25000 live births in Caucasians [1,2]. Recently, there has been substantial progress in the knowledge of CF as well as identification of more than 1,800 mutations in the CFTR gene that play a role in the phenotypic characterization of the disease. We describe a case of 2 Cystic Fibrosis siblings have markedly different clinical courses throughout their life. To objectively measure differences in their clinical courses, we reviewed clinical data points, chest x-rays and lung function tests [3].
Case Reports
Sibling 1
The patient is a 15-year-old white male, homozygous delta F508. His diagnosis with CF was at the age of 19 months exhibiting pulmonary exacerbations and cough manifestations and failure to thrive (FTT) with foul bulky stools. Upon investigating the etiology of the symptoms presented, a sweat chloride test was performed that yielded highly abnormal values, 105 and 109, indicating the subject was positive for cystic fibrosis. Confirmatory genetic testing was performed to reveal 2 copies of the F508del mutations. Stool testing revealed abnormal Pancreatic Fecal Elastase value of <15, this indicates he was completely pancreatic insufficient. Sibling 1 colonized respiratory MRSA at 3 years of age requiring hospital admission for intravenous antibiotics and comprehensive cystic fibrosis care. At 10 years of age Pseudomonas aeruginosa (PA) chronic colonization began.
Sibling 2
The patient is a 13-year-old white male, homozygous delta F508. Due to his sibling's confirmed diagnosis of CF, this subject was tested prenatally. It was confirmed via in utero genetic testing on amniotic fluid from amniocentesis sampling that he also was affected with CF having two copies of the F508del mutation. After birth tests confirmed, he too was fully pancreatic insufficient. Despite early intervention, his significant growth retardation from malabsorption of fat lead to surgical placement of a gastrostomy tube for overnight continuous high calorie and high fat supplementation and day time boluses. Furthermore, he received growth hormone prior to puberty.
Discussion
Hospitalizations
The variability in presentation of the CF disease continues when we compared the diagnosis for admission and hospitalization. Sibling 1 was admitted for either a pulmonary exacerbation or a sinus related procedure whereas sibling 2 was primarily admitted for gastrointestinal related complications and procedures.
Pulmonary function
There are a variety of measures used to assess the severity of the implications of CF on the lungs. The FEV1 is an excellent measure to represent the small and large airways and has been correlated to survival3.
Sibling 1 had an FEV1 that started at 110 percent predicted but currently averages around 90 percent predicted. This decline can be attributed to pulmonary exacerbations, chronic Pseudomonas aeruginosa, and MRSA colonization.
Sibling 1 FEV1 most recent 5-year trend was shown in Figure 1. Conversely sibling 2 remains stable. FEV1 values that persist within 110 percent predicted. Sibling 2 FEV1 most recent 5-year trend was shown in Figure 2.
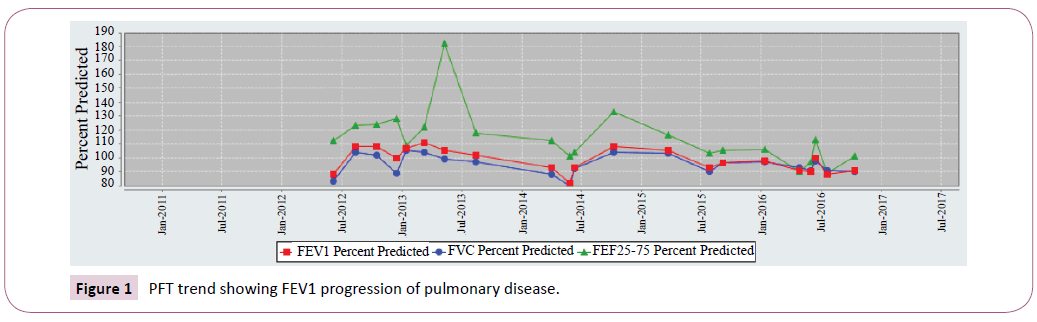
Figure 1: PFT trend showing FEV1 progression of pulmonary disease.
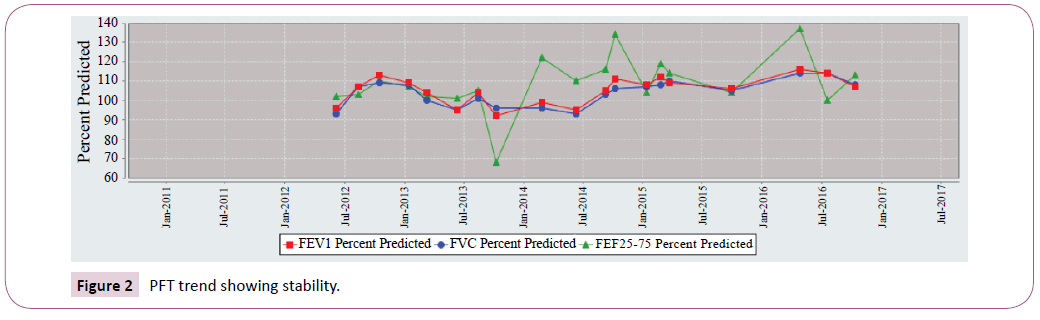
Figure 2: PFT trend showing stability.
Growth parameters
Sibling 1: At birth through 13 years of age his Body Mass Index (BMI) remained stable, 50th to the 70th percentile. Over a 3-year time period from age 12-16 years old, recurrent pulmonary exacerbations caused a marked transitory decline in BMI from the 20th to 30th percentile that was able to be recuperated.
Sibling 2: At birth plotted in the 60th percentile BMI and then markedly declined to under the 10th percentile. Even with supplemental G-tube nutritional supplements he has struggled to maintain a BMI between the 20th and 50th percentile (Table 1 and Figures 3 and 4).
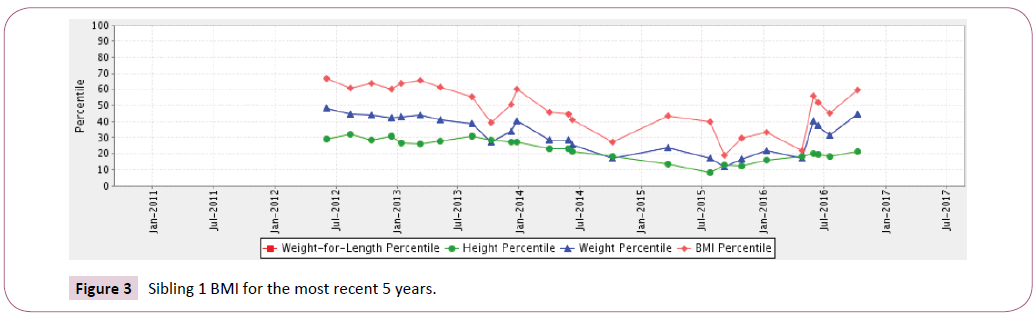
Figure 3: Sibling 1 BMI for the most recent 5 years.
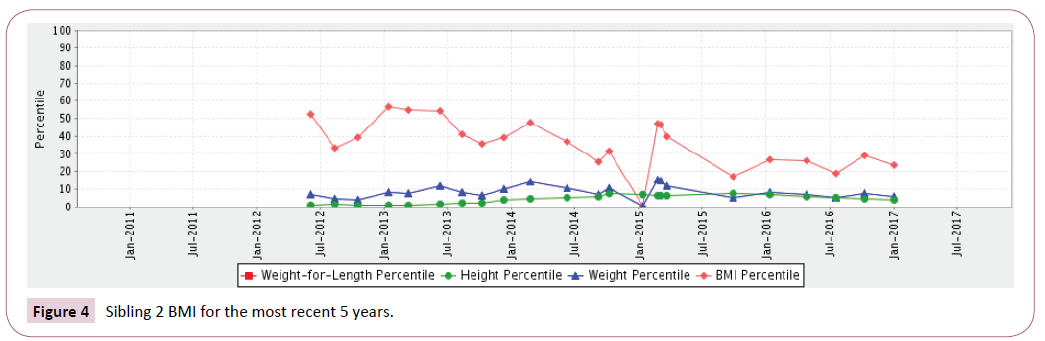
Figure 4: Sibling 2 BMI for the most recent 5 years.
| |
Sibling 1 |
Sibling 2 |
| Current age |
15-year-old, male |
13-year-old male |
| Age at diagnosis |
19 months |
In utero-amniocentesis |
| Sweat chloride test (mmol/L) |
105, 109 |
N/A |
| Respiratory disease |
Chronic sinusitis |
Chronic sinusitis, |
| FEV1% predicted |
89 |
100 |
| Airway colonization |
P. aeruginosa& MRSA |
P. aeruginosa |
| Pancreatic expression |
Pancreatic insufficiency |
Pancreatic insufficiency |
| Liver expression |
No |
No |
| Reproductive |
N/A |
N/A |
Table 1: Clinical expression in the two siblings affected by Cystic Fibrosis.
Conclusion
The purpose of this perspective case report is to focus attention to the intervariability of genetic expression in terms of phenotypic representation that can occur even within siblings. Clinicians and parents are encouraged to discuss the dichotomy of complications associated with the genetic components of CF, accentuating potential divergent clinical courses that necessitate a tailored approach to management. Genetic information is invaluable; nevertheless, it is not a reliable predictor for the spectrum of CF disease severity or clinical symptoms that will manifest.
Limitations
Some of the medical charts following the birth of the siblings were not located; however, we believe that we were able to present the whole medical history with no compromise.
19482
References
- Cutting GR (2010) Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann NY Acad Sci 1214:57-69.
- Scotet V, Dugueperoux I, Saliou P (2012) Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France. Orphanet J Rare Dis7:14.
- Kerem E, Reisman J, Corey M, Canny GJ, Levison H (1992) Prediction of mortality in patients with cystic fibrosis. N Engl J Med 326:1187-1191.

