Keywords
Epilepsy; Cognitive loss; Memory; Default mode network; Psychosis; Depression
Introduction
The flagship symptom of epilepsies is the epileptic seizure, but cognitive and behavioural changes, psychiatry symptoms and conditions intimately associate with them. We aim to present the putative mechanism of interictal epileptic psychiatric comorbidities, with special attention to cognitive changes. Because the temporo-limbic system has paramount importance in the regulation of emotional and mood-related functions as well as in the processes of memory and learning – cognition -, mesiotemporal lobe epilepsy out of the adulthood epilepsies, carries the highest risk for developing mental symptoms. Through its rich connections, the temporo-limbic system participates in the shaping of psychopathology symptoms of extra-temporal epilepsies as well; making mesiotemporal lobe epilepsy a proper model for studying the cognitive and mental comorbidities of epilepsies in general. Therefore, in our work we pay most attention to mesiotemporal lobe epilepsy.
Classification of mental comorbidities in epilepsy
The mental disturbances that are the complications of epilepsy belong to the group of ‘Organic, including symptomatic, mental disorders’ (ICD-10; F00-F09) [1]. In the classification system of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) [2], epilepsy-related disturbances fit into several additional groups: Group 20. ’Other clinically significant disturbances’ or group 17, ’Neurocognitive disturbances’. Childhood developmental epileptic encephalopathies may be consistent with DSM-5. 1. Neurodevelopmental disorders'. The depressive, psychotic, anxiety and compulsive disturbances belong to DSM5 2-6: 2. Schizophrenia spectrum and other psychotic disorders; 3. Depressive disorders; 4. Bipolar and related disorders; 5. Anxiety disorders; 6. Obsessive-compulsive and related disorders.
However, the epileptic variants of psychiatry conditions are usually different compared to the non-epileptic forms: they are shorter, non-familial and leave no mental deficits, contrasting schizophrenia. They may brittle from one type to another in one patient, carrying mixed features. Recognizing the atypical, epilepsy-specific presentation of some mental disorders, the International League against Epilepsy has built a classification system [3], discriminating typical versus atypical epileptic forms.
Based on the adoption of the existence of atypical forms by ILAE, we venture not to use detailed psychiatry nosology categories in our work; rather, we refer to groups based on leading symptoms such as depressions-mood disorders, anxiety syndromes, psychoses and schizophreniform psychoses in general.
The background of interictal psychopathology disturbances in epilepsy
While the cognitive damage of seizures and the instant harm of interictal discharges, called transient cognitive impairment, are relatively easy to interpret, the mechanism of enduring psychiatry symptoms and conditions far away from clinical seizures is less clear [4-8]. The following data demonstrate the proportions of such comorbidities.
More than half of the epilepsy population suffer from low moods and each epilepsy patient develops at least one episode of clinically significant depression, contrasting with just 16% of patients with diabetes, 17% with asthma and 9% of the general population [9-11]. Forty percent of epilepsy patients suffer with anxiety conditions [12]. The risk of suicide is elevated several- fold compared to the general population; twenty-fivefold in temporal lobe epilepsy [11,13-16]. The prevalence of psychosis is sevenfold of the general population, and the schizophreniform psychoses affect more than half of TLE patients [17-20]. During ten years of follow-up, 10% of children suffering in temporal lobe epilepsy have developed an episode of schizophrenic psychosis [21].
There are several concepts on the causes of chronic psychiatry comorbidity in epilepsy.
1. The unforeseen seizures maintaining uncertainty and defencelessness together with the complex psychosocial handicap related to epilepsy may have a psychopathology impact
2. Are the antiepileptic drugs responsible? Sometimes, but likely not essentially: we see the mental harm before introducing antiepileptic treatment and in spite of drug changes as well.
3. The interictal epileptiform activity causes chronic cognitive harm [22].
4. It has long been suspected that the abnormal sensorolimbic connections in mesiotemporal lobe epilepsy might cause pathologic hypersensitivity to external stimuli. Connectivity studies, especially those on resting state networks have confirmed such possibilities.
Ad 1. The unexpected seizures causing uncertainty and defenselessness may have a psychopathology impact
Epilepsy is a source of traumatic experiences, restrictions, defenselessness and stigmata. Most patients have social difficulties; are lonely and single. A seizure occurring in a community may be disgraceful, leading to embarrassment and repudiation, causing the isolation of the patient. Stigmatization may cause abnormal development of the self, and security restrictions increase shyness.
The repeated seizure-related loss of control is another specific traumatizing factor: experiencing doom may foster suicidal propensity and maladaptive strategies. Female gender, family stressors or the lack of family support are additional psychological risk factors.
Ad 2. The cognitive and psychopathology effects of antiepileptic drugs (AEDs)
Old AEDs as barbiturates and phenytoin typically cause psychopathology. Phenytoin and phenobarbitone are independent risk factors of suicide [23]. Vigabatrine, an irreversible gamma amino butyric acid (GABA)-transaminase inhibitor causes depression in 10% of patients [24], similarly to tiagabine; inhibiting GABA-uptake. Topiramate may aggravate depression [24]. Based upon one study; oxcarbazepine is an independent risk factor of depression; while other studies found its mood-improving effect [15,25,26]. Levetiracetam may cause irritability and fatigue, aggressive behaviour [27-29] and even psychosis [23,30]; 16% of patients treated with levetiracetam suffer psychiatry complications [31]. At the same time, due to its excellent antiepileptic properties, it is a good option in treating peri-ictal psychoses. Carbamazepine, lamotrigine and sodium valproate have an anti-depressive and mood stabilizing effect [24]. Lamotigine and gabapentine cause less mental adverse effects than the rest of AEDs [31]. Carbamazepine has a protective effect against psychosis [23].
The cognitive harm of AEDs correlates with the number administered in poly-therapy: the increase of AED-numbers given in combination correlates with an impairment of executive functions: “each additional drug matters” [32]. The excellent cognitive effects of lamotrigine and the less favorable ones of topiramate are well known. The cognitive spectrum of lacosamide is similar to LAM [33]. Levetiracetam improves the performance in visual memory and attention tests [27]. Only topiramate has caused any language-related functional network deactivation and dysphasia [34].
In summary, when dealing with mental changes in epilepsies, one needs to consider the potential impact of antiepileptic drugs, especially, if given in combination. Older antiepileptic drugs and topiramate may have cognitive harm, levetiracetam frequently compromises mood and might cause agitation, and lamotrigine is a mood-stabilizer sometimes also causing agitation. The drug interactions of AEDs (exceeding the frameworks if this review), especially with psychotropic drugs need special consideration when both types of drugs need to be administered together.
Ad 3. The chronic cognitive harm of interictal epileptiform activity: the memory impairment caused by spikes
Cognition has strong impact on apparently independent psychopathology conditions, even acting as their risk factors [35-37]. The memory process - encoding, consolidation and retrieval - may be compromised in several types of epilepsies; mildly and not perceived in some, or at a tragic and dramatic speed and extent in other ones e.g. in early childhood developmental encephalopathies.
The physiology of learning - encoding and consolidation of memory - associates to the hippocampi, the thalamo-cortical system and to nREM sleep [38-46].
Memory encoding
Based on animal experiments, the site of the activated CA1 and CA3 pyramidal cells is the presently known clue for the transcription from external stimulus to nervous signal, called encoding [47].
Synaptic plasticity
Learning depends on synaptic enhancement and weakening. Cellular learning, consistent with long-term potentiation is under the control of N-methyl-D-aspartate (NMDA)-receptors and other neurotransmitters. long-term potentiation [48] is a persistent strengthening of synapses based on recent patterns of activity, marked by the decrease of synaptic stimulusthreshold, the increase of the intensity and frequency of discharges, postsynaptic depolarization and the rate of calcium influx [42,43,49-52]. It develops due to repeated high frequency (tetanic) stimulation of a synapse, so that the involved neurons learn to convey a given stimulus.
Memory replay
One of the basic elements of memory consolidation [the stabilization of an engram] is the replay of daytime impulses. The incoming impulse activates the specific cortex, which in turn stimulates the granular cells of the hippocampus causing delayed activation of the hippocampal pyramidal cells during sleep and other off-line states of the brain [53]. The discharges of hippocampal pyramidal cells repeat the sequence of daytime impulses in an accelerated and condensed form; this is called replay [42,43,54-57]. The replay reinforces the unstable engrams and projects them to the frontal cortex, where they join stored memories and consolidate. e.g., the place-cells activate in a certain order at the time of the spatial orientationthen the sequence of these discharges reoccurs in the hippocampal pyramidal cells with a10-20-fold speed during the next sleeping period; consolidating ultimately in the cortex [53,54]. Thus, memory consolidation occurs in a corticohippocampo- cortical loop, broken if the hippocampus is damaged [43,55,56,58-60].
Sharp wave-ripple complexes
The key electrophysiology actor of memory-replay is the hippocampal sharp-wave ripple complex. It is made of a sharp wave arising from the excitatory system of the CA3 region [42,43,53-61] and an 80-200 Hz network ripple in the CA1 region [42]. The sharp-wave ripple complex emerges during off-line states: nREM sleep, resting and consummator periods [59-62]. Those studies where the abolishment of sharp-wave ripple complex hindered rats` space learning; have proven its essential role in memory consolidation [60,63].
The sharp-wave ripple complex and the interictal epileptic activity
Animal and human studies show that temporal epileptic spikes link with hippocampal ripples, suggesting that the sharp wave component of the complex suffers an epileptic derailment to an epileptic spike. This transcript (sham) of the sharp wave is unfit for memory consolidation [43,57-59,64,65]. In addition, the ripples of the complex may undergo an epileptic conversion as well, developing into pathological high frequency oscillations [43,66]. Thus, the epileptic spikes and pathological high frequency oscillations occupy the normal plastic process, making the system unserviceable [64,67]. Due to the sleep dependency of SPW-Rs, this conversion links to nREM sleep and favours after-learning periods.
Sharp-wave ripple complex and schizophrenias
In a schizophrenic, calcineurin-deficient mouse model (calcineurin is a forebrain specific phosphatase enzyme involved in synaptic plasticity) the hippocampal replay is abnormal: there are many sharp-wave ripple complexes, but their sequential pattern repeating daytime stimuli is absent. This links schizophrenia to cellular level synaptic dysfunction and cognitive harm [68,69].
The triad of slow waves, spindles and ripples in sleep
Within nREM sleep, sharp-wave ripple complexes link to neocortical slow (<1 Hz) waves, more so with the higher amplitude ones, emerging in the transition-zones of slow waves when the waves are turning from their up to their down states [61,70]. There is a coupling between sharp-wave ripple complexes and sleep spindles as well. In nREM sleep, those three patterns interlock under the direction of slow waves. The ripples of sharp-wave ripple complexes, carrying the reactivated engrams, fit into the sleep spindles feeding the hippocampal information into the neocortical networks, resulting in persistent synaptic enhancement called learning [61,71,72].
The homeostatic control of sleep slow waves
Sleep slow waves are under homeostatic control. Their number increases in the site of, and proportionally to the presleep exertion of the brain; then it decreases again after fulfilling the task [73-75]: recovering the exhausted synapses facilitated by pre-sleep daytime exertion. The name of this recovery process with slow wave decay overnight is synaptic downscaling; refreshing the saturated synapses and making them workable again. After a hard requisition, our sleep is “deeper”, containing more slow waves [41]. Slow waves are abundant in periods of high sleep-pressure (the first sleep cycle of the night, deep slow wave sleep and cyclic alternating pattern (CAP) A1), then they attenuate overnight [72,74-76].
Slow waves and spikes
The slow wave-tie of sharp-wave ripple complexes and their propensity to an epileptic derailment involve, that spikes and pathological high frequency oscillations couple with sleep slow waves [56,67,77-79]. Therefore, the highest spike-density is due in the periods of high homeostatic pressure during sleep, correlating with pre-sleep cortical requisition [77-80].
The spikes “mimic” engrams
The epileptic derailment of sharp-wave ripple complexes results in “senseless” spikes and pathological high frequency oscillations: Due to the spikes` similarity to the sharp waves of sharp-wave ripple complexes, the residual normal plasticity “mistakes” daytime spikes for memory traces. The condensed sequence of pre-sleep interictal spike-discharges could be detected during the subsequent sleep period over wide cortical regions meaning that spikes are replayed and processed, as normal engrams are [66,81]. Thus, spikes “behave” as normal engrams, enhancing the homeostatic pressure: in animals, seizures and spikes cause synaptic facilitation; and in human epilepsy patients, the sleep delta power correlates with the pre-sleep spike-number [64,82]. The spikes are dysfunctional in another way, as well, inhibiting the physiological homeostatic restitution. A high daytime spikenumber was followed by less slow-wave decay than normal learning during the subsequent sleep period [66]. In encephalopathy with electrical status epilepticus in sleep encephalopathy with status epilepticus in sleep (ESES), the continuous interictal discharges in sleep inhibited night-time slow wave decay, reducing next day’s learning capacity [73,82].
Summary: The multiple ways of memoryimpairment caused by interictal spikes
• During sleep, spikes and pathological high-frequency oscillations, as dysfunctional shams of sharp-wave ripple complexes are unfit to replay and consolidate normal memory (Figure 1).
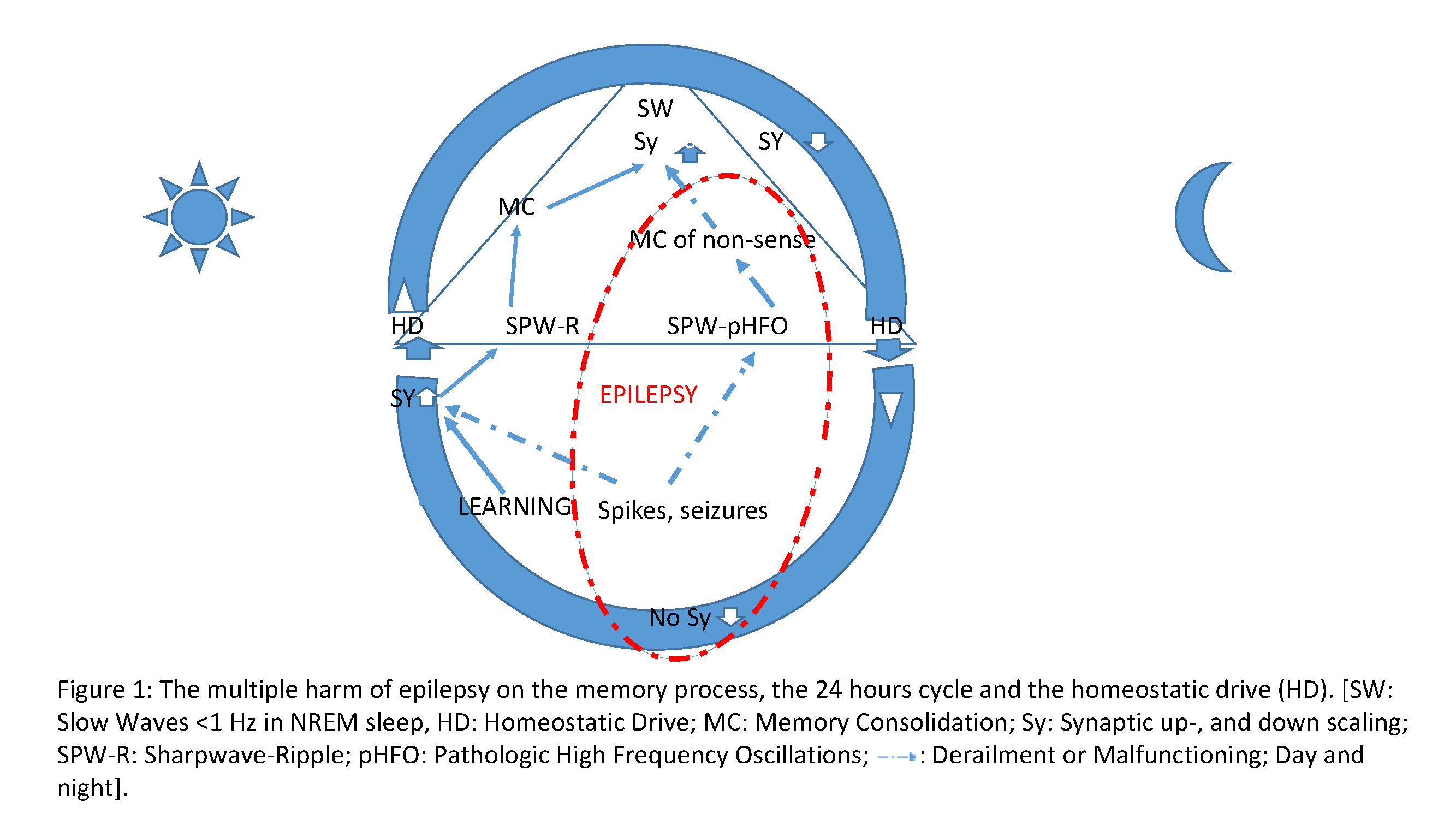
Figure 1: The multiple harm of epilepsy on the memory process, the 24 hours cycle and the homeostatic drive (HD). [SW: Slow Waves <1 Hz in NREM sleep, HD: Homeostatic Drive; MC: Memory Consolidation; Sy: Synaptic up-, and down scaling; SPW-R: Sharpwave-Ripple; pHFO: Pathologic High Frequency Oscillations;  : Derailment or Malfunctioning; Day and night].
: Derailment or Malfunctioning; Day and night].
• Daytime spikes behave as engrams: annexing synaptic capacity, they obstruct the elaboration of normal engrams.
• While daytime spikes cause homeostatic slow wave increase in sleep, they block slow wave decay overnight thus decreasing synaptic receptivity the day after.
• The distortion of the default mode network in mesiotemporal lobe epilepsy interlocks with spiking.
Clinical evidence on the harm of interictal spikes
Children: The prevalence of epilepsy is 30% in children with learning difficulties, and epileptic children without any IQ deficit may perform more poorly than controls in distinct learning domains, which justifies screening for such weaknesses in epileptic children [83,84]. The mood disorders seen in 34% of epileptic children can simulate cognitive deficits [85].
Sleep-related spikes and spike-wave pattern found in nonepileptic children is associated with cognitive deficits and behavioural changes, and the decrease of spike numbers resulted in behavioural improvement [86,87].
In mixed-type childhood epilepsies, attention- and shortterm memory deficits were seen [88]. The information processing and visuo-motor integration was poorer in those with interictal epileptic activity covering at least 10% of the daytime EEG [89].
In age-related childhood epilepsies, behavioural anomalies and cognitive loss [90,91], language and short-term memory deficits were detected [92-94], e.g. in benign centro-temporal epilepsy, proportionally with spiking the recognition of scary faces was deficient [95]. In another group of idiopathic focal epilepsy children, the memory consolidation related to nREM sleep correlated negatively with spiking [96-98].
The most severe forms of epilepsy-related cognitive loss are seen in the epileptic encephalopathies [82,99-101] with abundant epileptic activity during sleep. Electrical status epilepticus in nREM sleep presenting with global or patchy cognitive deterioration and its focal variant Landau-Kleffner syndrome with acquired aphasia are specific sleep-dependent forms [102-107]. No seizures or just sparse ones may occur with them; proving the independent harm of sleep related interictal activity [104]. Without the orienting lead of clinical seizures, these conditions cannot be diagnosed except with sleep EEGs [105-107].
Thirty percent of children with epilepsies suffer with attention-deficit hyperactivity syndrome. Both attentiondeficit hyperactivity syndrome and autism spectrum disorder link with epilepsy, as shown by the presence of interictal discharges carried by the affected non-epileptic children; epilepsy is an “autism sibling comorbidity disorder” [108-110].
Adults: The prevalence of dementia in epilepsy varies in a wide range: 8-17% [84]. Mesiotemporal lobe epilepsy may importantly compromise memory, negatively correlating with hippocampal volume and interictal activity [111-117]. The most affected field is executive functioning including verbal fluency and IQ [22]. “Everyday” memory - phone-numbers and dates - is impaired [114].
The role of interictal epileptic activity in the pathogenesis of dementia of adult neurodegenerative conditions, e.g. Alzheimer’s disease, has been raised [116,117].
Clinical consequences
The severe cognitive complications of interictal epileptic activity during nREM sleep [97,118-120] call attention to the lack of and need for a “spike-killing” treatment. Sleep EEG is indispensable in epileptology practice in general, and especially in searching for the cause of mental deterioration of childhood with no clinical seizures [76,80].
Ad 4. Aberrant sensoro-limbic connections in the mechanism of psychiatry comorbidities
Background: Brain functioning is related to the co-activation of interconnected nodes, outlining brain networks. The whole of functional and structural brain networks is called ‘connectome’ [analogue wording ‘genome’]; it is modelled by sophisticated mathematical methods [121-123]. The functional networks flexibly change their patterns depending on the context e.g. state, age, mood, pains etc., mutually influencing each other [123,124]. Somatic diseases also transform them; and their conversions are causes and markers of mental conditions [125-130]. The recognition of brain networks accompanies the development of the networkconcept of epilepsies and mental conditions [131-133]; e.g. taking into account the long reach of mesiotemporal lobe epilepsy involving wide spread bilateral regions; not just one mesio-temporal focus [105,134].
Methodology: functional MRI, diffusion tensor imaging, tractography: Functional connectivity is the term describing the connected [in time and frequency] activation of brain nodes in a network. EEG-based sophisticated calculations and functional MRI allow the modelling and imaging of brain networks [135]. Functional MRI traces out the regional oxygen consumption [BOLD signal- blood-oxygen-level dependent contrast imaging], which is proportional with the metabolism of a region scrutinized [136]. Thus, functional MRI can localise those regions co-activated by a task, allowing the mapping of the connected regions, constituting a functional network [137-139].
MRI tractography, based on diffusion tensor imaging [DTI] is the tool of mapping structural connectivity: MRI tractography images the neural pathways and tracts in the white matter, anatomically interconnecting the nodes [140]. DTI measures the diffusion of water molecules through white-matter bundles; which is necessarily slower across high-density structures. Its measure is anisotropic diffusion, depending on the thickness and wholeness of myelin [141].
The default mode network (DMN): We have learned the networks of elementary somatosensory functions [142], and those of sophisticated mental activities [143]. The latter are strongly related to the task-negative networks activating during resting states and inhibited by tasks; which had seemed artefacts -noises- initially [144-147]. The pattern of those networks related to attention [146], reward [147], mathematics [148] etc. determine our moods, level of anxiety, thinking and pathologic mental brain conditions.
The default mode network is one of the resting state networks, involved in the brain's floating, non-specific -defaultactivity [145,149]. Out of all other networks, the concordance of functional and structural connectivity is the strongest in this one [150], the “neurology base of self" [151]; related to a wide range of cognitive, mood-related and emotional functions.
Its main nodes are the posterior cingulate cortex and precuneus, the medial prefrontal cortex, the angular gyrus, the temporo-parietal junction, the temporal pole and hippocampus, the retrosplenial cortex and the posterior inferior pariet "https://en.wikipedia.org/wiki/Parietal_lobe"al lobe [149,152-155].
Epileptic networks: Epilepsy spreads across self-generating tracks - epileptic networks- in the brain producing new spikes, seizures and additional epileptic regions (foci) on its way [156-158]. In this sense, epilepsies are progressive conditions. Typically, mesiotemporal epilepsy spreads to the contralateral hippocampus, the homo-lateral temporal neocortex and the ipsilateral frontal lobe [104,159].
Interaction between interictal epileptic activity and the default mode network: Immediately before spikes occur, there is an increase of default mode network-activity, while spikes associate with a decrease of the activity of several default mode network nodes [138], suggesting an interaction [137,160]. This mutual effect might explain why in encephalopathy with electrical status epilepticus in sleep the spiking regions show an increased default mode network activity, while outside the epileptogenic area, it deactivates pathologically [82].
Abnormal functional connectivity in mesiotemporal lobe epilepsy: There are several epilepsy-specific functional connectivity changes [161]. The network changes persist after seizure freedom is reached, i.e. chronic epilepsy leads to a permanent network distortion (and likely related mental symptoms); making early treatment vital [162]. The pattern of functional connectivity changes helps to discriminate mesiotemporal epilepsy cases with or without hippocampal sclerosis, right and left, poor and good prognosis, easy- and difficult-to-treat ones [163-170] and those ones developing after-, versus those without a febrile convulsion (Table 1).
| Mesiotemporal epilepsy |
Schizophreniform psychosis |
Depression/bipolar disorder |
| Decreased C between H and DMN [163-165] |
Decreased C between H and DMN [187] |
Decreased medial prefrontal cortex C [186] |
| Hyperconnetivity between H and DMN [166] Increased C between the occipital and cingulate cortex [134,171] |
DMN hyper-, [132,187] or hypo- connectivity [155,180,188]. |
Decreased DMN C in the left caudate nucleus, right anterior cingulate cortex, right angular gyrus, bilateral medial prefrontal cortex and right praecuneus [190]. |
| C changes between H-DMN and H-SMA correlating [180,188] with positive symptoms. |
Increased C between the DMN and the subgenual region [192, 193] |
| C changes in the bilateral praecuneus and right inferior parietal lobulus [180]. |
|
| Increased neocortico-limbic C [193] |
Distorted cortico-subcortical networks: thalamocortical, frontolimbic and cortico-cerebellar [194-197]. |
Inter-hemispheric and limbic C changes; increased amygdala-medial praefrontal C [186] |
| Hippocampo-limbic C changes correlating with the depressive symptoms positively in left mesiotemporal epilepsy, negatively in right mesiotemporal epilepsy [198]. |
C changes in hearing and language networks, in the frontopolar network, in basal ganglia [199]. |
|
| Changes of C in amygdalar emotional face recognition networks, mainly in right temporal epilepsy [167,168] |
changes of C between nucleus accumbens-DMN, and cingulo-opercular network [201] |
|
| Increased intra-H C [134, 171] |
Abnormal intra-H C [189] |
|
Table 1: Network changes shared by mesiotemporal epilepsy and non-epileptic psychoses (schizophrenia and schizophrenia spectrum disorders -ICD10-5 F20-29; DSM5-2; depressions and bipolar disorders -ICD10-5 F30-39; DSM5-3,4) (C= connectivity, H= hippocampus, hippocampal, DMN: default mode network, SMA: Supplementary sensory-motor area).
The default mode network undergoes a transformation in mesiotemporal epilepsy. There is an intra-hippocampal increase of connectivity and a decrease of connectivity between the hippocampus and the default mode network [171-180]. The limbic-neocortical connectivity is low as well (Table 1).
The functional connectivity changes found in non-epileptic psychoses and affective conditions are similar to those in epilepsy: There are default mode network excitation/ inhibition changes and imbalance in psychotic and affective disorders, affecting mentally intact relatives as well [180-185]. Although due to methodology issues, the data on network changes are hard to summarize there is growing evidence on the change of the default mode network or some of its nodes in non-epileptic mental conditions.
In Table 1 and Figure 2, we present those connectivity changes strikingly shared by non-epileptic psychoses and epilepsies, to highlight the similarities, which are in line with structural changes and might contribute to the development of psychoses in epilepsy.
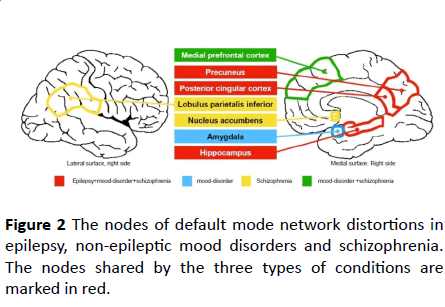
Figure 2: The nodes of default mode network distortions in epilepsy, non-epileptic mood disorders and schizophrenia. The nodes shared by the three types of conditions are marked in red.
Epilepsy causes structural connectivity changes: There are unexpected structural changes in non-lesional epilepsies [164-172]. The volume of white matter decreases in childhood epilepsies. In surgical tissue samples of mesiotemporal epilepsy patients, there were significantly more excitatory and inhibitory neurones both in the white and grey matter, than in non-epileptic controls [173]. There were white matter abnormalities in the fornix, the cingulate gyrus, the corpus callosum and the left superior frontal gyrus with widespread extra-temporal grey matter atrophy despite excess aberrant neurogenesis [174-177]. In the default mode network pathways of animas modelling mesiotemporal epilepsy, the axonal calibres were decreased and the myelin-sheets abnormal [151] and in humans, the degree of the anterior temporal, uncinate and lower longitudinal fasciculus abnormalities correlated with the severity of mesiotemporal epilepsy [173].
In summary, because the degree of white matter impairment correlates with the frequency of partial seizures and the duration of mesiotemporal epilepsy, the causative role of epilepsy seems likely [186-200].
How does epilepsy cause white matter damage? The overuse of epileptic networks by spiking and seizures may not be responsible in itself, because generally, the excess use of brain systems - practice - improves functioning: e.g., vision, movements, thinking etc. do not cause harm; rather, they improve performance and lead to the anatomic enlargement of the involved system, contrasting epilepsy.
Structural connectivity-changes in non-epileptic psychoses and mood disorders (ICD10-5 F20-29; DSM5-2; ICD10-5- F30-39; DSM5-3,4): Schizophrenias develop white matter disorganisation. Schizophrenia and bipolar disorders share frontal white matter anomalies, more marked in the former [186]. The fronto-temporal pathways are abnormal early, even in the prodromal phase and in adolescents. The decrease of white matter volume correlates with the duration of the disease [140].
There are grey matter volume changes as well, affecting the frontal, temporal and anterior cingulate cortices [200,201]. The volume-increase of the thalamus, and the decrease of the left middle and superior frontal gyrus, significantly link with cognitive deficits [194,195].
In summary, it is possible that epileptic psychoses and mood disorders are related to the structural changes caused by epilepsy, partially resembling those seen in genuine mental conditions.
Summary of the chapter ‘Aberrant sensoro-limbic connections in the mechanism of psychiatry comorbidities’: Epilepsy, especially mesiotemporal epilepsy
• Changes the default mode network.
• It causes structural damage affecting both grey and white matter especially the tracts and nodes of the default mode network.
These network changes and structural alterations resemble those seen in genuine, major psychiatry conditions probably contributing to the appearance of their atypical forms in epilepsies.
Summary Considerations
Mechanism of psychopathology in epilepsy
Depressions and epilepsy: Psychosocial factors do certainly affect patients’ mood and some of the antiepileptic medications may contribute as well, but the remarkably frequent depressive co-morbidity compared to the rest of chronic conditions suggests an additional biological background of depression in epilepsy [202-206]. There are pathogenic factors shared by depressions and epilepsies e.g. the overlapping functional and structural connectivity changes [207-212]. The harm of temporal lobe epileptic activity exerts on plastic functions is another major contributing factor [35-37,212,213] while conversely, depression impairs memory [214].
In epileptic animal models, low levels of serotonin, noradrenaline, dopamine and GABA promote the production of new epileptic areas - epileptic kindling- increasing seizure frequency and disease severity. Some antidepressants may inhibit or reverse this [209,215,216].
Higher serotonin levels allow long lasting long-term potentiation in response to external stimuli, while lower ones support the replay of engram [54]: the serotonergic disturbances related to depression have multi-lateral impacts on memory consolidation. The triangle of epilepsy, depression and cognitive loss interact, mutually augmenting each other [9,215,217].
Epilepsy and psychoses: Based on the frequently seen interictal psychoses in patients with complex partial seizures, Gibbs and Gibbs [218] presumed the existence of common mesio-temporal mechanisms. Several data support this suspicion.
• Connectivity studies [134,219]: epilepsies lead to atypical psychotic disorders, though distorting the default mode network.
• There are shared genetic factors of temporal lobe epilepsy and schizophrenia [220].
• In surgical samples of TLE patients suffering also in psychoses or major depression, the accumulation of neuroinflammatory molecules resembled those in non-epileptic psychiatry conditions discriminating temporal lobe epilepsy with or without a psychosis [216].
• We have known since Kraepelin and Bleuler that schizophrenia associate with cognitive harm, even before the actual onset of psychosis. The cognitive loss is not a complication of psychosocial factors and occurs in the normal relatives as well; it might be a risk factor and harbinger of poor prognosis in schizophrenias [221].
Acknowledgements
The authors are thankful for the support of the National Brain Research Program 2017-1.2.1-NKP-2017-00002.
22034
References
- ICD-10 (2010) International Statistical Classification of Diseases and Related Health Problems: 10th Revision (ICD-10) version.
- Diagnostic and Statistical Manual of Mental Disorders (2014) American Psychiatric Association (5th edn).
- Krishnamoorthy ES, Trimble MR, Blumer D (2007) Classification of psychiatric disorders of epilepsy according to the proposal of the ILAE Commission on Psychobiology of Epilepsy. Epilepsy Behav 10: 349-353.
- Aldenkamp J (2004) Effects of epileptiform EEG discharges on cognitive function: Is the concept of “transient cognitive impairment” still valid? Epilepsy Behav 5: 25-34.
- Binnie CD, Marston D (1992) Cognitive correlates of interictal discharges. Epilepsia 6: S11-17.
- Mameniskiene R, Jatuzis D, Kaubrys G, Budrys V (2006) The decay of memory between delayed and long-term recall in patients with temporal lobe epilepsy. Epilepsy Behav 8: 278-288.
- Siebelink BM, Bakker DJ, Binnie CD, Nolst K, Trenité DG (1988) Psychological effects of subclinical epileptiform EEG discharges in children. II. General intelligence tests. Epilepsy Res 2: 117-121.
- Postnikova TY, Zubareva OE, Kovalenko AA, Kim KK, Magazanik LG, et al. (2017) Status Epilepticus Impairs Synaptic Plasticity in Rat Hippocampus and Is Followed by Changes in Expression of NMDA Receptors. Biochemistry (Mosc) 82: 282-290.
- Nogueira MH, Yasuda CL, Coan AC, Kanner AM, Cendes F (2017) Concurrent mood and anxiety disorders are associated with pharmacoresistant seizures in patients with MTLE. Epilepsia 58: 1268-1276.
- Błaszczyk B, Czuczwar SJ (2016) Epilepsy coexisting with depression. Pharmacol Rep 68: 1084-1092.
- Blümer D, Montouris G, Davies K, Wyler A, Phillips B, et al. (2002) Suicide in epilepsy: Psychopathology, pathogenesis, and prevention. Epilepsy Behav 3: 232-241.
- Tram P, Sauro KM, Scott BP, Wiebe S, Fiest KM, et al. (2017) The prevalence of anxiety and associated factors in persons with epilepsy. Epilepsia 58: e107-e110.
- Thurman DJ, Logroscino G, Beghi E, Hauser WA, Hesdorffer DC, et al. (2017) The burden of premature mortality of epilepsy in high-income countries: A systematic review from the mortality task force of the international league against epilepsy. Epilepsia 58: 17-26.
- Tian N, Cui W, Zack M, Kobau R, Fowler KA, et al. (2016) Suicide among people with epilepsy: A population-based analysis of data from the U.S. National Violent Death Reporting System, 17 states, 2003-2011. Epilepsy Behav 61: 210-217.
- Mula M, Sander JW (2013) Suicide risk in people with epilepsy taking antiepileptic drugs. Bipolar Disord 15: 622-627.
- Harris EC, Barraclough B (1997) Suicide as an outcome for mental disorders. A meta-analysis. Br J Psychiatry 170: 205-228.
- Kanner AM, Rivas-Grajales AM (2016) Psychosis of epilepsy: A multifaceted neuropsychiatric disorder. NS Spectr 21: 247-257.
- Slater E, Beard AW, Glithero E (1963) The schizophrenialike psychoses of epilepsy. Br J Psychiatry 109: 95-150.
- Mendez MF, Grau R, Doss RC, Taylor JL (1993) Schizophrenia in epilepsy: Seizure and psychosis variables. Neurology 43: 1073-1077.
- Trimble MR, Schmitz B (2004) The psychoses of epilepsy psychiatric comorbidity in epilepsy: Basic mechanisms, diagnosis and treatment. McConnell HW, Snyder PJ. (Eds) (1st edn) American Psychiatric Press Inc, USA. pp: 169-186.
- Lecardeur L, Meunier-Cussac S, Dollfus S (2013) Cognitive deficits in first episode psychosis patients and people at risk for psychosis: from diagnosis to treatment. Encephale. 39: 64-71.
- Dinkelacker V, Xin X, Baulac M, Samson S, Dupont S (2016) Interictal epileptic discharge correlates with global and frontal cognitive dysfunction in temporal lobe epilepsy. Epilepsy Behav 62: 197-203.
- Kalinin VV (2007) Suicidality and antiepileptic drugs: is there a link? Drug Saf 30: 123-142.
- Rajna P (2007) Effects of antiepileptic drugs on mood of people with epilepsy. (Az antiepileptikumok hatása az epilepsziás betegek kedélyállapotára). Ideggyógy Sz 60: 381-391.
- Bosak M, Turaj W, Dudek D, Siwek M, Szczudlik A (2015) Depressogenic medications and other risk factors for depression among Polish patients with epilepsy. Neuropsychiatr Dis Treat 11: 2509-2517.
- Grimaldi-Bensouda L, Nordon C, Rossignol M, Jardon V, Boss V, et al. (2017) Antiepileptic drugs and risk of suicide attempts: a case-control study exploring the impact of underlying medical conditions. Pharmacoepidemiol Drug Saf 26: 239-247.
- Schoenberg MR, Rum RS, Osborn KE, Werz MA (2017) A randomized, double-blind, placebo-controlled crossover study of the effects of levetiracetam on cognition, mood, and balance in healthy older adults. Epilepsia 58:1566-1574.
- Hurtado B, Koepp MJ, Sander JW, Thompson PJ (2006) The impact of levetiracetam on challenging behavior. Epilepsy Behav 8: 588-592.
- Dinkelacker V, Dietl T, Widman G, Lengler U, Elger CE (2003) Aggressive behavior of epilepsy patients in the course of levetiracetam add-on therapy: report of 33 mild to severe cases. Epilepsy Behav 4: 537-547.
- Shukla G, Gupta A, Agarwal P, Poornima S (2016) Behavioral effects and somnolence due to levetiracetam versus oxcarbazepine - a retrospective comparison study of North Indian patients with refractory epilepsy. Epilepsy Behav 64: 216-218.
- Weintraub D, Buchsbaum R, Resor SR, Hirsch LJ (2007) Psychiatric and behavioral side effects of the newer antiepileptic drugs in adults with epilepsy. Epilepsy Behav 10: 105-110.
- Witt JA, Elger CE, Helmstaedter C (2015) Adverse cognitive effects of antiepileptic pharmacotherapy: Each additional drug matters. Eur Neuropsychopharmacol 25: 954-959.
- Helmstaedter C, Witt JA (2013) The longer-term cognitive effects of adjunctive antiepileptic treatment with lacosamide in comparison with lamotrigine and topiramate in a naturalistic outpatient setting. Epilepsy Behav 26: 182-187.
- Wandschneider B, Burdett J, Townsend L, Hill A, Thompson PJ, et al. (2017) Effect of topiramate and zonisamide on fMRI cognitive networks. Neurology 88: 1165-1171.
- Bora E, Murray RM (2014) Meta-analysis of cognitive deficits in ultra-high risk to psychosis and first-episode psychosis: Do the cognitive deficits progress over, or after, the onset of psychosis? Schizophrenia Bull 40: 744-755.
- Barch DM, Sheffield JM (2014) Cognitive impairments in psychotic disorders: common mechanisms and measurement. World Psychiatry 13: 224-232.
- Kandratavicius L, Ruggiero RN, Hallak JE, Garcia-Cairasco N, Leite JP (2012) Pathophysiology of mood disorders in temporal lobe epilepsy. Rev Bras Psiquiatr 34: S233-245.
- Lee KA, Wilson MA (2002) Memory of sequential experience in the hippocampus during slow wave sleep. Neuron 36: 1183-1194.
- Hasson U, Chen J, Honey CJ (2015) Hierarchical process memory: Memory as an integral component of information processing. Trends Cog Neurosci 19: 304-313.
- Wilson MA, McNaughton BL (1994) Reactivation of hippocampal ensemble memories during sleep. Science 265: 676-679.
- Born J (2010) Slow-wave sleep and the consolidation of long-term memory. World J Biol Psychiatry 11: 16-21.
- Buzsáki G (2015) Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25: 1073-1188.
- Buzsáki G (1989) Two-stage model of memory trace formation: A role for "noisy" brain states. Neuroscience 31: 551-570.
- Lee JL, Everitt BJ, Thomas KL (2004) Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 304: 839-843.
- Rolls ET (1996) A theory of hippocampal function in memory. Hippocampus 6: 601-620.
- Chen Z, Wilson MA (2017) Deciphering neural codes of memory during sleep. Trends Neurosci 40: 260-275.
- Kouvaros S, Papatheodoropoulos C (2016) Theta burst stimulation-induced LTP: Differences and similarities between the dorsal and ventral CA1 hippocampal synapses. Hippocampus 26: 1542-1559.
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39.
- Bliss T, Lømo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232: 331-356.
- Morris RG, Anderson E, Lynch GS, Baudry M (1986) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319: 774-776.
- Morris RG (1989) Synaptic plasticity and learning: selective impairment of learning rats and blockade of long-term potentiation in vivo by the N-methyl-D-aspartate receptor antagonist AP5. J Neurosci 9: 3040-3057.
- Jahnke S, Timme M, Memmesheimer RM (2015) A unified dynamic model for learning, replay, and sharp-wave/ripples. J Neurosci 35: 16236-16258.
- Haq RU, Anderson ML, Hollnagel JO, Worschech F, Sherkheli MA, et al. (2016) Serotonin dependent masking of hippocampal sharp wave ripples. Neuropharmacology 101: 188-203.
- Rothschild G, Eban E, Frank LM (2017) A cortical–hippocampal–cortical loop of information processing during memory consolidation. Nature Neurosci 20: 251-259.
- Clemens Z, Janszky J, Szűcs A, Békésy M, Clemens B, et al. (2003) Interictal epileptic spiking during sleep and wakefulness in mesial temporal lobe epilepsy: a comparative study of scalp and foramen ovale electrodes. Epilepsia 44: 186-192.
- Carr MF, Jadhav SP, Frank LM (2011) Hippocampal replay in the awake state: a potential substrate for memory consolidation and retrieval. Nature Neurosci 14: 147-153.
- Karlsson MP, Frank L (2009) Awake replay of remote experiences in the hippocampus. Neuroscience 12: 913-918.
- McClelland JL, Goddard NH (1996) Considerations arising from a complementary learning systems perspective on hippocampus and neocortex. Hippocampus 6: 654-665.
- Roux L, Hu B, Eichler R, Stark E, Buzsáki G (2017) Sharp wave ripples during learning stabilize the hippocampal spatial map. Nat Neurosci 20: 845-853.
- Clemens Z, Mölle M, Erőss L, Barsi P, Halász P, et al. (2007) Temporal coupling of parahippocampal ripples, sleep spindles and slow oscillations in humans. Brain 130: 2868-2878.
- Liotta A, Caliskan G, Haq RU, Hollnagel JO, Rosler A, et al. (2011) Partial disinhibition is required for transition of stimulus-induced sharp wave-ripple complexes into recurrent epileptiform discharges in rat hippocampal slices. J Neurophysiol 105: 172-187.
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB (2009) Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci 12: 1222-1223.
- Gelinas JN, Khodagholy D, Thesen T, Devinsky O, Buzsáki G (2016) Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nature Med 22: 641-648.
- Pinto LF, Adda CC, Silva LC, Banaskiwitz NH, Passarelli V, et al. (2017) Contralateral interictal and ictal EEG epileptiform activity accentuate memory impairment in unilateral mesial temporal sclerosis patients. Neuropsychology 31: 268-276.
- Boly M, Jones B, Findlay G, Plumley E, Mensen A, et al. (2017) Altered sleep homeostasis correlates with cognitive impairment in patients with focal epilepsy. Brain 140: 1026-1040.
- Jiruska P, Alvarado-Rojas C, Schevon CA, Staba R, Stacey W, et al. (2017) Update on the mechanisms and roles of high-frequency oscillations in seizures and epileptic disorders. Epilepsia 58:1330-1339.
- Suh J, Foster DJ, Davoudi H, Wilson MA, Tonegawa S (2013) Impaired hippocampal ripple-associated replay in a mouse model of schizophrenia. Neuron 80: 484-493.
- O'Donnell P (2013) Of mice and men: What physiological correlates of cognitive deficits in a mouse model of schizophrenia tell us about psychiatric disease. Neuron 80: 265-266.
- Frauscher B, Von Ellenrieder N, Ferrari-Marinho T, Avoli M, Dubeau F, et al. (2015) Facilitation of epileptic activity during sleep is mediated by high amplitude slow waves. Brain 138: 1629-1641.
- Mölle M, Born J (2011) Slow oscillations orchestrating fast oscillations and memory consolidation. Prog Brain Res 193: 93-110.
- Wei Y, Krishnan GP, Bazhenov M (2016) Synaptic mechanisms of memory consolidation during sleep slow oscillations. J Neurosci 36: 4231-4247.
- Bölsterli HBK, Fattinger S, Kurth S, Lebourgeois MK, Ringli M, et al. (2014) Spike wave location and density disturb sleep slow waves in patients with CSWS (continuous spike waves during sleep). Epilepsia 55: 584-591.
- Tononi G, Cirelli C (2003) Sleep and synaptic homeostasis: A hypothesis. Brain Res Bull 62: 143-150.
- Tononi G, Cirelli C (2014) Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81: 12-34.
- Halász P (2013) How sleep activates epileptic networks? Epilepsy Res Treat 2013: 425697.
- Rossi GF, Colicchio G, Pola P (1984) Interictal epileptic activity during sleep: A stereo-eeg study in patients with partial epilepsy. Electroenceph Clin Neurophysiol 58: 97-106.
- Sammaritano M, Gigli GL, Gotman J (1991) Interictal spiking during wakefulness and sleep and the localization of foci in temporal lobe epilepsy. Neurology 41: 290-297.
- Malow BA, Lin X, Kushwaha R, Aldrich MS (1998) Interictal spiking increases with sleep depth in temporal lobe epilepsy. Epilepsia 9: 1309-1316.
- Horváth A, Szűcs A, Barcs G, Kamondi A (2017) Sleep EEG detects epileptiform activity in Alzheimer’s disease with high sensitivity. J Alz Dis 56: 1175-1183.
- Bower MR, Kucewicz MT, St Louis EK, Meyer FB, Marsh WR, et al. (2017) Reactivation of seizure-related changes to interictal spike shape and synchrony during postseizure sleep in patients. Epilepsia 58: 94-104.
- Filippini M, Arzimanoglou A, Gobbi G (2013) Neuropsychological approaches to epileptic encephalopathies. Epilepsia 54: 38-44.
- Subota A, Pham T, Jetté N, Sauro K, Lorenzetti D, et al. (2017) The association between dementia and epilepsy: A systematic review and meta-analysis. Epilepsia 58: 962-972.
- Fastenau PS, Shen J, Dunn DW, Austin JK (2008) Academic underachievement among children with epilepsy: Proportion exceeding psychometric criteria for learning disability and associated risk factors. J Learning Disabilities 41: 195-207.
- Paradiso S, Hermann BP, Blumer D, Davies K, Robinson RG (2001) Impact of depressed mood on neuropsychological status in temporal lobe epilepsy. JNNP 70: 180-185.
- Capdevila OS, Dayyat E, Kheirandish-Gozal L, Gozal D (2008) Prevalence of epileptiform activity in healthy children during sleep. Sleep Med 9: 303-309.
- Ronit M, Pressler RM, Robinson RO, Binnie CD (2005) Treatment of interictal epileptiform discharges can improve behavior in children with behavioral problems and epilepsy. J Pediatrics 146: 112-117.
- Noeker M, Haverkamp F (1988) Pathways to psychopathology in epilepsy. Psychosomatics. 29: 295-300.
- Ebus S, Arends J, Hendriksen J, Van der Horst E, De la Parra N, et al. (2012) Cognitive effects of interictal epileptiform discharges in children. Eur J Paediatr Neurol 16: 697-706.
- Scott CM (2013) The relationship between sleep-activated interictal epileptiform discharges and intelligence in children. Neurodiagn J 53: 289-311.
- Kwon S, Seo HE, Hwang SK (2012) Cognitive and other neuropsychological profiles in children with newly diagnosed benign rolandic epilepsy. Korean J Pediatr 55: 383-387.
- Verly M, Gerrits R, Lagae L, Sunaert S, Rommel N, et al. (2017) Evaluation of the language profile in children with rolandic epilepsy and developmental dysphasia: Evidence for distinct strengths and weaknesses. Brain Lang 170: 18-28.
- Neumann H, Helmke F, Thiels C, Polster T, Selzer LM, et al. (2016) Cognitive development in children with benign rolandic epilepsy of childhood with centrotemporal spikes - Results of a current systematic database search. Fortschr Neurol Psychiatr 84: 617-632.
- Verrotti A, Filippini M, Matricardi S, Agostinelli MF, Gobbi G, et al. (2014) Memory impairment and benign epilepsy with centrotemporal spike (BECTS): a growing suspicion. Brain Cogn 84: 123-131.
- Ciumas C, Laurent A, Saignavongs M, Ilski F, De Bellescize J, et al. (2017) Behavioral and fMRI responses to fearful faces are altered in benign childhood epilepsy with centrotemporal spikes (BCECTS). Epilepsia 58: 1716-1727.
- Galer S, Urbain C, De Tiège X, Emeriau M, Leproult R, et al. (2015) Impaired sleep-related consolidation of declarative memories in idiopathic focal epilepsies of childhood. Epilepsy Behav 43: 16-23.
- Urbain C, Di Vincenzo T, Peigneux P, Van Bogaert P (2011) Is sleep-related consolidation impaired in focal idiopathic epilepsies of childhood? A pilot study. Epilepsy Behav 22: 380-384.
- Chan S, Pressler R, Boyd SG, Baldeweg T, Cross JH, et al. (2017) Does sleep benefit memory consolidation in children with focal epilepsy? Epilepsia 58: 456-466.
- Lado FA, Rubboli G, Capovilla G, Avanzini G, Moshé SL, et al. (2013) Pathophysiology of epileptic encephalopathies. Epilepsia 54: 6-13.
- Von Deimling M, Helbig I, Marsh ED (2017) Epileptic encephalopathies-Clinical syndromes and pathophysiological concepts. Curr Neurol Neurosci Rep 17: 10.
- Shbarou R, Mikati MA (2016) The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin Pediatr Neurol 23: 134-142.
- Chen XQ, Zhang WN, Hu LY, Liu MJ, Zou LP, et al. (2016) Syndrome of electrical status epilepticus during sleep: Epileptic encephalopathy related to brain development. Pediatr Neurol 56: 35-41.
- Gencpinar P, Dundar NO, Tekgul H (2016) Electrical status epilepticus in sleep (ESES)/continuous spikes and waves during slow sleep (CSWS) syndrome in children: An electroclinical evaluation according to the EEG patterns. Epilepsy Behav 61: 107-111.
- Halász P, Szűcs A (2017) Epilepsy, sleep and cognitive impairment. Elsevier, . Amsterdam, The Netherlands.
- Pal DK, Ferrie CP, Addis L, Akiyama T, Capovilla G, et al. (2016) Idiopathic focal epilepsies: the "lost tribe". Epileptic Disord 18: 252-288.
- Halász P, Kelemen A, Clemens B, Saracz J, Rosdy B, et al. (2005) The perisylvian epileptic network. A unifying concept. Ideggyogy Sz 58: 21-31.
- Hegyi M, Siegler Z, Fogarasi A, Barsi P, Halász P (2016) Long-term follow-up of lesional and non-lesional patients with electrical status epilepticus in slow wave sleep. Ideggyógyászati Sz 69: 21-28.
- Ghacibeh GA, Fields C (2015) Interictal epileptiform activity and autism. Epilepsy Behav 47: 158-162.
- Besag F, Gobbi G, Caplan R, Dunn DW, Sillanpää M, et al. (2016) Psychiatric and Behavioural Disorders in Children with Epilepsy (ILAE Task Force Report): Epilepsy and ADHD. Epileptic Disord 47: 119-125.
- Diaz-Beltran L, Esteban FJ, Varma M, Ortuzk A, David M, et al. (2017) Cross-disorder comparative analysis of comorbid conditions reveals novel autism candidate genes. BMC Genomics 18: 315.
- Zhao F, Kang H, You Li, Rastogi P, Venkatesh D, et al. (2014) Neuropsychological deficits in temporal lobe epilepsy: A comprehensive review. Ann Indian Acad Neurol 17: 374-382.
- Harden CL (2007) New evidence supports cognitive decline in temporal lobe epilepsy. Epilepsy Currents 7: 12-14.
- Tavakoli M, Barekatain M, Doust HTN, Molavi H, Nouri RK, et al. (2011) Cognitive impairments in patients with intractable temporal lobe epilepsy. J Research in Med Sci 16: 1466-1472.
- Rzezak P, Lima EM, Gargaro AC, Coimbra E, De Vincentiis S, et al. (2017) Everyday memory impairment in patients with temporal lobe epilepsy caused by hippocampal sclerosis. Epilepsy Behav 69: 31-36.
- Pacagnella D, Lopes TM, Morita ME, Yasuda CL, Cappabianco FA, et al. (2014) Memory impairment is not necessarily related to seizure frequency in mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 55: 1197-1204.
- Cretin B, Philippi N, Dibitonto L (2017) Epilepsy at the prodromal stages of neurodegenerative diseases. Geriatr Psychol Neuropsychiatr 5: 75-82.
- Horváth A, Szűcs A, Barcs G, Noebels JL, Kamondi A, et al. (2016) Epileptic seizures in Alzheimer’s disease: A review. Alz Dis Assoc Disord 30: 186-192.
- Barry JM, Holmes GL (2016) Why are children with epileptic encephalopathies encephalopathic? J Child Neurol 31: 1495-1504.
- Holmes GL, Lenck-Santini PP (2006) Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav 8: 504-515.
- Holmes GL (2014) What is more harmful, seizures or epileptic EEG abnormalities? Is there any clinical data? Epileptic Disord 1: S12-S22.
- Hagmann P (2005) From diffusion MRI to brain connectomics (Thesis). Lausanne: EPFL.
- Sporns O, Tononi G, Kötter R (2005) The human connectome: A structural description of the human brain. PLoS Computational Biology 1: e42.
- Cabral J, Vidaurre D, Marques P, Magalhães R, Moreira PS, et al. (2017) Cognitive performance in healthy older adults relates to spontaneous switching between states of functional connectivity during rest. Scientific Rep 7: 1.
- Soares JM, Marques P, Magalhães R (2017) The association between stress and mood across the adult lifespan on default mode network. Brain Struc Func 222: 101.
- Bomyea J, Matthews SC, Buchsbaum MC, Spadoni AD, Strigo IA, et al. (2017) Neural Differences Underlying Face Processing in Veterans with TBI And Co-Occurring TBI and PTSD. J Affect Disord 223: 130-138.
- Lutz PE, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, et al. (2017) Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Am J Psychiatry.
- Mosher Val, Swain MG, Pang JXQ, Kaplan GG, Sharkey KA, et al. (2017) Primary biliary cholangitis alters functional connections of the brain's deep gray matter. Clin Transl Gastroenterol 8: E107.
- Browndyke JN, Berger M, Harshbarger TB, Smith PJ, White W, et al. (2017) Resting-state functional connectivity and cognition after major cardiac surgery in older adults without preoperative cognitive impairment: Preliminary findings. J Am Geriatr Soc 65: E6-E12.
- Etkin A, Prater KE, Schatzberg AF, Menon V, Greicius MD (2009) Disrupted amygdalar subregion functional connectivity and evidence of a compensatory network in generalized anxiety disorder. Arch Gen Psychiatry 66: 1361-1372.
- Liao X, Vasilakos AV, He Y (2017) Small-world human brain networks: Perspectives and challenges. Neurosci Biobehav Rev 77: 286-300.
- Spencer SS (2002) Neural networks in human epilepsy: evidence of and implications for treatment. Epilepsia 43: 219-227.
- Peeters SC, Van de Ven V, Gronenschild EH (2011) Genetic Risk and Outcome of Psychosis (G.R.O.U.P.). Reduced cortical thickness as an outcome of differential sensitivity to environmental risks in schizophrenia. Biol Psychiatry 69: 487-494.
- Kumar A, Cook IA (2002) White matter injury, neural connectivity and the pathophysiology of psychiatric disorders. Dev Neurosci 24: 255-261.
- DeSalvo MN, Douw L, Tanaka N, Claus R, Steven MS (2014) Altered structural connectome in temporal lobe epilepsy. Radiology 270: 842-848.
- Snyder AZ, Raichle ME (2012) A brief history of the resting state: The Washington University perspective. Neuroimage 62: 902-910.
- Ogawa S, Lee TM, Kay AR, Tank DW (1990) Brain magnetic resonance imaging with contrast dependent on blood oxygenation. PNAS 87: 9868-9872.
- Lopes R, Moeller F, Besson P, Ogez F, Szurhaj W, et al. (2014) Study on the relationships between intrinsic functional connectivity of the default mode network and transient epileptic activity. Front Neurol 5: 201.
- Jacobs J, Menzel A, Ramantani G, Körbl K, Assländer J, et al. (2014) Negative BOLD in default-mode structures measured with EEG-MREG is larger in temporal than extra-temporal epileptic spikes. Front Neurosci 8: 335.
- Jäger V, Dümpelmann M, LeVan P, Georgia R, Irina M, et al. (2015) Concordance of epileptic networks associated with epileptic spikes measured by high-density EEG and Fast fMRI. PLoS One 10: e0140537.
- Guye M, Bartolomei F, Ranjeva JP (2008) Imaging structural and functional connectivity: Towards a unified definition of human brain organization? Curr Opin Neurol 21: 393-403.
- Landek-Salgado MA, Faust TE, Sawa A (2016) Molecular substrates of schizophrenia: homeostatic signalling to connectivity. Mol Psychiatry 21: 10-28.
- Avanzini P, Abdollahi RO, Sartori I, Caruana F, Pelliccia V, et al. (2016) Four-dimensional maps of the human somatosensory system. PNAS 113: 1936-1943.
- Lisman JE, Grace AA (2005) The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron 46: 703-713.
- Andrews-Hanna JR, Reidler JS, Huang C, Buckner RL (2010) Evidence for the default network’s role in spontaneous cognition. J Neurophysiol 104: 322-335.
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, et al. (2001) A default mode of brain function. PNAS 98: 676-682.
- de Schotten MT, Dell'Acqua F, Forkel SJ, Simmons A, Vergani F, et al. (2011) A lateralized brain network for visuospatial attention. Nature Neurosci 14: 1245-1246.
- Camara E, Rodriguez-Fornells A, Ye Z, Münte TF (2009) Reward networks in the brain as captured by connectivity measures. Front Neurosci 3: 350-362.
- Menon V, Mackenzie K, Rivera SM, Reiss AL (2002) Prefrontal cortex involvement in processing incorrect arithmetic equations: evidence from event-related fMRI. Hum Brain Mapp 16: 119-130.
- Greicius MD, Krasnow B, Reiss AL, Menon V (2003) Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proc Natl Acad Sci 100: 253-258.
- Horn A, Ostwald D, Reisert M (2014) The structural-functional connectome and the default mode network of the human brain. Neuroimage 1: 142-151.
- Andrews-Hanna, Jessica R (2012) The brain's default network and its adaptive role in internal mentation. Neuroscientist 18: 251-270.
- Khalsa S, Mayhew SD, Chechlacz M, Bagary M, Bagshaw AP (2014) The structural and functional connectivity of the posterior cingulate cortex: comparison between deterministic and probabilistic tractography for the investigation of structure-function relationships. Neuroimage 1: 118-127.
- Vatansever D, Menon DK, Manktelow AE, Sahakian BJ, Stamatakis EA (2015) Default mode dynamics for global functional integration. J Neurosci 35: 15254-15262.
- Andrews-Hanna JR, Smallwood J, Spreng RN (2014) The default network and self-generated thought: component processes, dynamic control, and clinical relevance. Annals of the New York Academy of Sciences 1316: 29-52.
- Anticevic A, Cole MW, Murray JD, Corlett PR, Wang XJ, et al. (2012) The role of default network deactivation in cognition and disease. Trends Cogn Sci 16: 584-592.
- Bartolomei F, Lagarde S, Wendling F, McGonigal A, Jirsa V, et al. (2017) Defining epileptogenic networks: Contribution of SEEG and signal analysis. Epilepsia 58: 1131-1147.
- Goddard GV (1967) Development of epileptic seizures through brain stimulation at low intensity. Nature 214: 1020-1021.
- Alkawadri R, Gonzalez-Martinez J, Gaspard N, Alexopoulos AV (2016) Propagation of seizures in a case of lesional mid-cingulate gyrus epilepsy studied by stereo-EEG. Epileptic Disord 18: 418-425.
- Centeno M, Carmichael DW (2014) Network connectivity in epilepsy: Resting state fMRI and EEG–fMRI contributions. Front Neurol 5: 93.
- Fahoum F, Zelmann R, Tyvaert L, Dubeau F, Gotman J (2013) Epileptic discharges affect the default mode network--FMRI and intracerebral EEG evidence. PLoS One 8: e68038.
- Pedersen M, Omidvarnia A, Curwood EK, Walz JM, Rayner G, et al. (2017) The dynamics of functional connectivity in neocortical focal epilepsy. NeuroImage Clin 15: 209-214.
- Maccotta L, Lopez MA, Adeyemo B, Ances BM, Day BK, et al. (2017) Postoperative seizure freedom does not normalize altered connectivity in temporal lobe epilepsy. Epilepsia 58: 1842-1851.
- Haneef Z, Lenartowicz A, Yeh HJ, Levin HS, Engel J Jr, et al. (2014) Functional connectivity of hippocampal networks in temporal lobe epilepsy. Epilepsia 55: 137-145.
- Min L, Boris CB, Seok-Jun H, Caldairou B, Bernasconi A, et al. (2016) Superficial white matter in temporal lobe epilepsy: a key link between structural and functional network disruptions. Brain 139: 2431-2440.
- James GA, Tripathi SP, Ojemann JG, Gross RE, Drane DL (2013) Diminished default mode network recruitment of the hippocampus and parahippocampus in temporal lobe epilepsy. J Neurosurg 119: 288-300.
- Steiger BK, Muller AM, Spirig E, Toller G, Jokeit H (2017) Mesial temporal lobe epilepsy diminishes functional connectivity during emotion perception. Epilepsy Res 134: 33-40.
- Stewart E, Catroppa C, Lah S (2016) Theory of mind in patients with epilepsy: A systematic review and meta-analysis. Neuropsychol Rev 26: 3-24.
- Realmuto S, Zummo L, Cerami C, Agrò L, Dodich A, et al. (2015) Social cognition dysfunctions in patients with epilepsy: Evidence from patients with temporal lobe and idiopathic generalized epilepsies. Epilepsy Behav 47: 98-103.
- Morgan VL, Englot DJ, Rogers BP, Landman BA, Cakir A, et al. (2017) Magnetic resonance imaging connectivity for the prediction of seizure outcome in temporal lobe epilepsy. Epilepsia 58: 1251-1260.
- Li H, Ji C, Zhu L, Huang P, Jiang B, et al. (2017) Reorganization of anterior and posterior hippocampal networks associated with memory performance in mesial temporal lobe epilepsy. Clin Neurophysiol 128: 830-838.
- Cataldi M, Avoli M, De Villers-Sidani E (2013) Resting state networks in temporal lobe epilepsy. Epilepsia 54: 2048-2059.
- Gross DW, Concha L, Beaulieu C (2006) Extratemporal white matter abnormalities in mesial temporal lobe epilepsy demonstrated with diffusion tensor imaging. Epilepsia 47: 1360-1363.
- Richter Z, Janszky J, Sétáló G Jr, Horváth R, Horváth Z, et al. (2016) Characterization of neurons in the cortical white matter in human temporal lobe epilepsy. Neuroscience 333: 140-150.
- Kreilkamp BA, Weber B, Richardson MP, Keller SS (2017) Automated tractography in patients with temporal lobe epilepsy using TRActs Constrained by UnderLying anatomy (TRACULA). Neuroimage Clin 14: 67-76.
- Miró J, Gurtubay-Antolin A, Ripollés P, Sierpowska J, Juncadella M, et al. (2015) Interhemispheric microstructural connectivity in bitemporal lobe epilepsy with hippocampal sclerosis. Cortex 67: 106-121.
- Chang YA, Kemmotsu N, Leyden KM, Kucukboyaci NE, Iragui VJ, et al. (2017) Multimodal imaging of language reorganization in patients with left temporal lobe epilepsy. Brain Lang 170: 82-92.
- Leyden KM, Kucukboyaci NE, Puckett OK, Lee D, Loi RQ, et al. (2015) What does diffusion tensor imaging (DTI) tell us about cognitive networks in temporal lobe epilepsy? Quant Imaging Med Surg 5: 247-263.
- Scanlon C, Mueller SG, Cheong I, Hartig M, Weiner MW, et al. (2013) Grey and white matter abnormalities in temporal lobe epilepsy with and without mesial temporal sclerosis. J Neurol 260: 2320-2329.
- Parent JM (2007) Adult neurogenesis in the intact and epileptic dentate gyrus. Prog Brain Res 163: 529-540.
- Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, et al. (2017) Searching for cross-diagnostic convergence: Neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry 10: 848-861.
- Garrity AG, Pearlson GD, McKiernan K, Lloyd D, Kiehl KA, et al. (2007) Aberrant “default mode” functional connectivity in schizophrenia. Am J Psychiatry 164: 450-457.
- Fox JM, Abram SV, Reilly JL, Eack S, Goldman MB, et al. (2017) Default mode functional connectivity is associated with social functioning in schizophrenia. J Abnorm Psychol 126: 392-405.
- Du Y, Pearlson GD, Lin D, Sui J, Chen J, et al. (2017) Identifying dynamic functional connectivity biomarkers using GIG-ICA: Application to schizophrenia, schizoaffective disorder, and psychotic bipolar disorder. Human Brain Mapping 38: 2683-2708.
- Bassett DS, Bullmore ET (2016) Small-world brain networks revisited. Neuroscientist.
- Karbasforoushan H, Woodward ND (2012) Resting-state networks in schizophrenia. Curr Top Med Chem 12: 2404-2414.
- O'Donoghue S, Holleran L, Cannon DM, McDonald C (2017) Anatomical dysconnectivity in bipolar disorder compared with schizophrenia: A selective review of structural network analyses using diffusion MRI. J Affect Disord 209: 217-228.
- Mothersill O, Tangney N, Morris DW, McCarthy H, Frodl T, et al. (2016) Further evidence of alerted default network connectivity and association with theory of mind ability in schizophrenia. Schizophr Res S0920-9964: 30553-30559.
- Liu Y, Zhang Y, Lv L, Wu R, Zhao J, et al. (2017) Abnormal neural activity as a potential biomarker for drug-naive first-episode adolescent-onset schizophrenia with coherence regional homogeneity and support vector machine analyses. Schizophr Res S0920-9964: 30215-30223.
- Samudra N, Ivleva EI, Hubbard NA, Rypma B, Sweeney JA, et al. (2015) Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Res 233: 148-157.
- Liu F, Hu M, Wang S, Guo W, Zhao J, et al. (2012) Abnormal regional spontaneous neural activity in first-episode, treatment-naive patients with late-life depression: a resting-state fMRI study. Prog Neuropsychopharmacol Biol Psychiatry 39: 26-31.
- Hamilton JP, Farmer M, Fogelman P, Gotlib IH (2015) Depressive rumination, the default-mode network, and the dark matter of clinical neuroscience. Biol Psychiatry 8: 224-230.
- Posner J, Hellerstein DJ, Gat I, Mechling A, Klahr K, et al. (2013) Antidepressants normalize the default mode network in patients with dysthymia. JAMA Psychiatry 70: 373-382.
- Luo C, Qiu C, Guo Z, Fang J, Li Q, et al. (2012) Disrupted functional brain connectivity in partial epilepsy: A resting-state fMRI study. PLoS ONE 7: e28196.
- Skåtun KC, Kaufmann T, Doan NT, Alnæs D, Córdova-Palomera A, et al. (2016) Consistent functional connectivity alterations in schizophrenia spectrum disorder: A multisite study. Schizophr Bull 43: 914-924.
- Skåtun KC, Kaufmann T, Brandt CL, Doan NT, Alnæs D, et al. (2017) Thalamo-cortical functional connectivity in schizophrenia and bipolar disorder. Brain Imaging Behav.
- Knöchel C, Stäblein M, Prvulovic D, Ghinea D, Wenzler S, et al. (2016) Shared and distinct gray matter abnormalities in schizophrenia, schizophrenia relatives and bipolar disorder in association with cognitive impairment. Schizophr Res 171: 140-148.
- Sheffield JM, Barch DM (2016) Cognition and resting-state functional connectivity in schizophrenia. Neurosci Biobehav Rev 61: 108-120.
- Kemmotsu N, Kucukboyaci NE, Cheng CE, Girard HM, Tecoma ES, et al. (2013) Alterations in functional connectivity between the hippocampus and prefrontal cortex as a correlate of depressive symptoms in temporal lobe epilepsy. Epilepsy Behav 29: 552-559.
- Oh J, Chun JW, Kim E, Park HJ, Lee B, et al. (2016) Aberrant neural networks for the recognition memory of socially relevant information in patients with schizophrenia. Brain Behav 7: e00602.
- Sharma A, Wolf DH, Ciric R, Kable JW, Moore TM, et al. (2017) Common dimensional reward deficits across mood and psychotic disorders: A connectome-wide association study. Am J Psychiatry 174: 657-666.
- Liberg B, Rahm C, Panayiotou A, Pantelis C (2016) Brain change trajectories that differentiate the major psychoses. Eur J Clin Invest 46: 658-674.
- Takayanagi M, Wentz J, Takayanagi Y, Schretlen DJ, Ceyhan E, et al. (2013) Reduced anterior cingulate gray matter volume and thickness in subjects with deficit schizophrenia. Schizophr Res 150: 484-490.
- Vaughan DN, Raffelt D, Curwood E, Tsai MH, Tournier JD, et al. (2017) Tract-specific atrophy in focal epilepsy: Disease, genetics, or seizures? Ann Neurol 81: 240-250.
- Olney JW, Collins RC, Sloviter RS (1986) Excitotoxic mechanisms of epileptic brain damage. Adv Neurol 44: 857-877.
- Nagy SA, Horváth R, Perlaki G, Orsi G, Barsi P, et al. (2016) Age at onset and seizure frequency affect white matter diffusion coefficient in patients with mesial temporal lobe epilepsy. Epilepsy Behav 61: 14-20.
- Lacey CJ, Salzberg MR, D'Souza WJ (2015) Risk factors for depression in community-treated epilepsy: A systematic review. Epilepsy Behav 43: 1-7.
- Kanner AM, Soto A, Gross-Kanner H (2004) Prevalence and clinical characteristics of postictal psychiatric symptoms in partial epilepsy. Neurology 62: 708-713.
- Kanner AM (2016) Management of psychiatric and neurological comorbidities in epilepsy. Nat Rev Neurol 12: 106-116.
- Mao Q, Gong X, Zhou C, Tu Z, Zhao L, et al. (2017) Up-regulation of SIRT6 in the hippocampus induced rats with depression-like behavior via the block Akt/GSK3β signaling pathway. Behav Brain Res 323: 38-46.
- Dubé C (2007) Neuropeptide Y: Potential role in recurrent developmental seizures. Peptides 28: 441-446.
- Chen T, Kendrick KM, Wang J, Wu M, Li K, et al. (2017) Anomalous single-subject based morphological cortical networks in drug-naive, first-episode major depressive disorder. Hum Brain Mapp.
- Rayner G (2017) The contribution of cognitive networks to depression in epilepsy. Epilepsy Curr 17: 78-83.
- Kandratavicius L, Peixoto-Santos JE, Monteiro MR, Scandiuzzi RC, Carlotti CG, et al. (2015) Mesial temporal lobe epilepsy with psychiatric comorbidities: a place for differential neuroinflammatory interplay. J Neuroinflammation 12: 38.
- Leal SL, Noche JA, Murray EA, Yassa MA (2017) Disruption of amygdala-entorhinal-hippocampal network in late-life depression. Hippocampus 27: 464-476.
- Bagdy G, Kecskeméti V, Riba P, Jakus R (2007) Serotonin and epilepsy. J Neurochemistry 100: 857-873.
- Wittner L, Maglóczky Z (2017) Synaptic Reorganization of the Perisomatic Inhibitory Network in Hippocampi of Temporal Lobe Epileptic Patients. Biomed Res Int.
- Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, et al. (1999) Reduced cortical g-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry 56: 1043-1047.
- Gibbs FA, Gibbs EL (1952) Atlas of electroencephalography. Addison-Wesley Press; Cambridge, MA, USA.
- Di Lorenzo G, Daverio A, Ferrentino F, Santarnecchi E, Ciabattini F, et al. (2015) Altered resting-state EEG source functional connectivity in schizophrenia: the effect of illness duration. Front Human Neuroscience 9: 234.
- Cascella NG, Schretlen DJ, Sawa A (2009) Schizophrenia and epilepsy: Is there a shared susceptibility? Neurosci Res 63: 227-235.
- Reichenberg A (2005) Cognitive impairment as a risk factor for psychosis. Dialogues Clin Neurosci 7: 31-38.
- Stip E (2006) Cognition, schizophrenia and the effect of antipsychotics. Encephale 32: 341-50.

