INTRODUCTION
|
| Coronaviruses (CoV) are enveloped single-stranded positive sense RNA viruses infect human and wide variety of animals and birds causing severe illness. Coronaviruses that infect humans include human coronavirus 1 (alfa), 2 (beta) and 3 (gamma). The severe acute respiratory syndrome (SARS) has been associated with Betacornovirus, which include four distantly related subgroups. It has been proved that the severe onset of SARS epidemic in 2003 with over 800 fatalities was evoluted in a market of lifecaged dogs market in China. Studies confirmed that caged animals and bats were the potential intermediate hosts [1, 2]. |
| The dangerous fate of coronaviruses arises from its ability of high recombination frequencies. The unique viral replication, the low fidelity of coronavirus- encoded polymerases and high recombination allows for unexpected viral evolution to infect other hosts, more clinical deficits and resistance to therapy or vaccination. SARS-CoV is a model virus for studying viral mutation, recombination and evolution. The human virus (SARS-CoV OC43) was thought to be evoluted from bovine coronavirus. Furthermore, several reports assured the changes in tissue tropism by evolution of a new porcine respiratory corona virus pathogen from a gastrointestinal ancestor [2, 3]. Such changes in host and tissue tropism elucidate the possibility of emerging new viruses and occurrence of new diseases manifestations associated with the unpredictable fates of coronavirus evoluting sequence. |
| In September 2012, the Middle East Respiratory syndrome Coronavirus (MERS-CoV) was initially identified in Saudi Arabia [4]. Following these first record positive cases were then recorded in United Kingdom, Germany, Tunisia, Jordan, Italy, Qatar, Emirates and France [5-8]. Saudi Arabia is the most heavily impacted with MERS-CoV, recoding more than 50% fatality rate by showing 38 deaths out of 64 cases [8]. |
| Among known viruses, coronavirus has a large genome of more than 27 kb. The genome of coronavirus encodes 23 putative proteins including 4 major structural proteins; nucleocapsid [N protein], spike [S protein], membrane [M] and small envelop proteins [E]. The variations is S protein among coronaviruses is associated different species specificity, serological response and tissue tropism. The S protein is a glycoprotein essential for viral attachment to the cell surface receptors and translocation into the infected cells. The S protein is cleaved in host cells into S1 and S2 subunits. S1 protein binds the host receptor, while S2 receptor mediates membrane fusion [9]. In this report, the full genome sequence of MERSCoV was compared with other CoV sequences. Furthermore, the highly variable spike region was also retrieved and analyzed. The phylogenetic relations and its indication are thoroughly discussed. |
Materials and Methods
|
|
Retrieval of full genome sequences
|
| The full-length CoV genomes were retrieved from the nucleotides depository at the National Center for Biotechnology Information (NCBI). |
|
Search for putative domains
|
| The full genome sequence of MERS-CoV was analyzed for all potential domains by the domain search tools at NCBI. The sequence for spike protein was identified and used to BLAST the protein database for knowing the high homology hits. |
|
Multiple sequence alignment
|
| Multiple sequence alignment was constructed by using Clustal omega tool at the European Bioinformatics Institute. The output file was retrieved and manually edited by GeneDoc software. The similarity and homology percent was calculated by Ugene 1.12.2 for mac. |
|
Phylogenetic analysis
|
| The output alignments from Clustal omega were further investigated by Dendroscope software for creation of phylogenetic trees. The phylogenetic tree was obtained by neighbor-joining method in output formats of radial phylogram or circular calidogram. |
Results and discussion
|
| Bioinformatics is a useful tool in analysis of new genes, proteins or whole genome. Annotations and analysis of sequences is a gold standard in the finding of new functions, drug targets and analysis of recently emerged infectious diseases [10-15]. To identify the distinguishing feature of the new MERS-CoV, the full genome sequence of MERS-CoV as well as the sequence of S protein was retrieved, compared to other CoV full genome sequences and analyzed by bioinformatics tools. |
|
Comparison of Coronaviruses whole genome
|
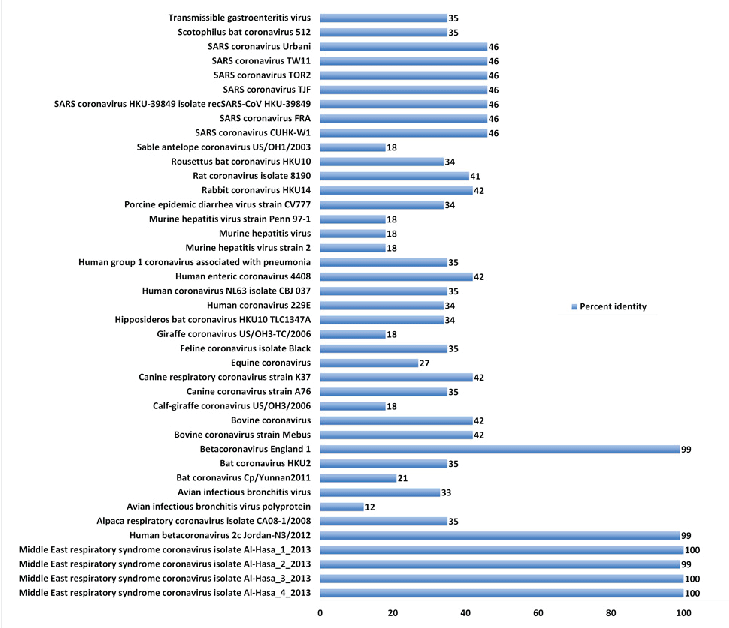
| About 41 full genome sequences representing different isolates of coronaviruses were retrieved from the genome database (Table 1, Fig. 1). In general, low to medium identity was evident in comparing MERS-CoV with other CoVs. Taking the four Middle East isolates from Al-Hasa as a reference, the following conclusions can be elucidated. First, it shares about 46% homology with the SARS coronavirus genome sequence. Second, it shares low identity with CoV isolates from animal origins e.g. Sable antelope (18%), rabbit (42%), murine hepatitis (18%), giraffe (18%), bovine (42%), canine (35%), avian infectious bronchitis (12%) and alpaca (35%). Lastly, 34-35%, identity with isolates of CoV from bats. |
| This result highlights the genetic variability of CoV. While keeping low identities, the clinical signs and virus replication is still sharing common mechanisms. Phylogenetic of the whole genome. |
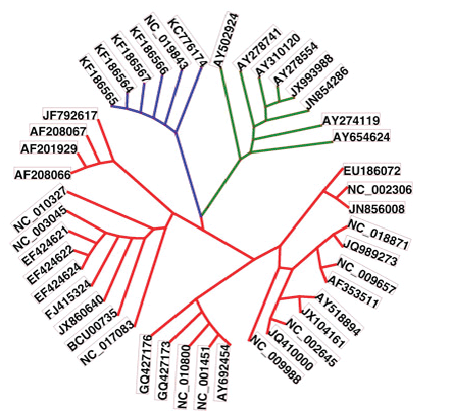
| The phylogenetic relations of the retrieved full genome sequences are shown in Fig. 2. The MERSCoVs isolated from Al-Hasa and their highly relevant isolated in Jordan and England were shown in blue. The highly related sequences were highlighted in green. From the figure, MERS-CoV isolates were highly related to SARS CoV isolates and those of Asian origin. |
|
Is MERS-CoV a SARS-CoV?
|
| The clinical signs of MERS-CoV include fever, acute respiratory distress and in some cases renal failure [4, 5]. These signs are more or less similar to the signs of SARS-CoV. Based on molecular bases, the new MERS-CoV is not SARS-CoV. MERS-CoV does not use the SARS-CoV receptor during entry into the cells. Furthermore, the mode of binding of MERS-CoV with the cell receptors is different from SARS CoV [16]. Phylogenetic maps showed that both MERS-CoV and SARS-CoV are sharing at least 40% of their sequences. |
|
Phylogenetic relations of S protein
|
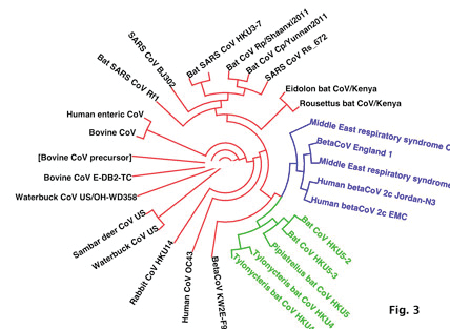
| Analysis of the Spike protein of different CoV sequences reveals perfect homology of MERS-CoV sequences from Saudi Arabia with related isolates from England and Jordan. This group of isolates is highly related to bat-derived CoV HKU4 and HKU5 (Table 2, Fig. 3). In contrast, it is highly divergent from other bat-derived CoV as Eidolon, Rousettus, Rhinoloplus bats isolates. Interestingly, MERS-CoV was showed poor relations from CoV from animal sources as Samber deer, bovine, waterbuck and rabbit coronaviruses. This underscores the role animals as carriers for MERS-CoV and highlights the potential incriminations of bats in the initiation of MERS-CoV. |
| The spike protein sequences show more or less low identity among different coronaviruses, showing as low as 12%. S protein is therefore, a region, which is highly variable among coronaviruses that lead to recognition of different cellular receptors and altered pathogenicity. The low homology of MERSCoV S protein with the other S proteins reveals a significant change in the molecular interaction of the new virus with host receptors. |
|
Zoonotic or human-human transmission of MERS-CoV
|
| Zoonotic exposure implies the transmission of the diseases through contact with animals, in this case as cattle, bats, or cats infected with coronavirus. Until the moment the role of reservoirs or intermediate hosts of MERT-CoV is still not understood. By using phylogenetic analysis, the possible way of transmission of MERS-CoV was deduced from the phylogenetic relation of the newly evolved virus with the previous sequences isolated from bats and other animals and human. |
| A limited human-human transmission was suggested by the analysis of surveillance data in France. The first 2 MERS-CoV-reported cases were in contact together in the hospital for 4 days, however, in a total number of 201 individuals were in contact with 2 cases only 2 were positive and the others were asymptomatic [7]. |
| Camels are thought to be the source for MERS-CoV [17]. Recently, neutralizing antibodies against MERSCoV were found in all samples of camels in the Middle East [18]. This does not suggest that camels are the source of infection. Confirmatory tests and isolation of the virus from camels is still yet undetermined. Based on the obtained phylogenetic tree, poor relation was found between MERS-CoV and those coronaviruses isolated from bovines and other animals. Bovine CoV was found to be circulating in cattle causing unnoticed mild symptoms to sever outbreaks. Furthermore, the virus was involved in enteric manifestations and now evolved to include also respiratory symptoms [19-21]. Camels are infected with CoV-like viruses (genetically related with bovine CoV) causing enteric symptoms in calves [20, 22]. However, the presence or absence of respiratory forms of CoV in camels is still not known. The claim that camels might be the source of MERS-CoV still needs further investigations. |
| Bats are the major reservoir for CoV infections. Recently, 96 bats representing 7 species (Rhinopoma hardwickii, Rhinopoma microphyllum, Taphozous perforatus, Pipistrellus kuhlii, Eptesicus bottae, Eidolon helvum, and Rosettus aegyptiacus) were captured and examined for their CoV contents [23]. The prevalence of CoVs was high (≈28% of fecal samples), MERS-CoV was found in only 1 bat. MERS positive signal was obtained in PCR analysis of the T. perforatus bat captured in Bisha near the home and workplace of the MERS index case-patient. This result indicates that bats are the ultimate source MERS-CoV. To this end, more details about the origin, carriers, intermediate hosts and pathogenesis of MERS-CoV still needs further investigations. |
Tables at a glance
|
 |
|
| Table 1 |
Table 2 |
|
| |
Figures at a glance
|
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
|
| |
|
|