The diversity of TBI
The incidence of TBI in Canada is estimated to be 100 injuries per day with the death rate being on the order of 50,000 people per year (1). Motor vehicle crashes are the most common cause of TBI in the civilian population (2). Sports injuries represent major sources of injury in children, adolescents and young adults (3) and seniors are vulnerable to falls (4). TBI is also considered the “signature injury” in modern warfare, as approximately 10-20% of veterans returning from the Iraq or Afghanistan wars have suffered a TBI, 80% of which are due to blast-related diffuse axonal injury (5).
TBI severity is classified on a continuum from mild to severe according to the presence of mental status changes, amnesia, loss of consciousness (LOC), neurological deficits and anatomical lesions (6). Several scales have been used to assist in the clinical classification of TBI severity, with the Glasgow Coma Scale (GCS) (7) remaining the most widely used. The GCS evaluates the best eye, verbal and motor responses to a stimulus, and the score of each individual element as well as the total GCS score is important. The lowest possible GCS score is 3 and signifies deep coma, whereas the highest is 15 and indicates a fully awake, responsive individual. Severe head injuries have a GCS from 3-8, moderate head injuries have a GCS from 9-12 and mild head injuries have a GCS from 13-15.
While classification of moderate and severe brain injuries is relatively straightforward, there is considerable diversity in how mild traumatic brain injuries (mTBI) are recognized and reported. Historically, mild head injury was defined as trauma to the head with a GCS greater than 12, LOC of less than 20 min, hospitalization less than 48h and no evidence of lesion upon neuroimaging (8). In 1993, the American Congress of Rehabilitation Medicine (ACRM) developed criteria to define mTBI that also included alteration of consciousness at the time of incident and posttraumatic amnesia of less than 24h, to recognize that subjects with mTBI can exhibit persistent emotional, cognitive, behavioral and physiological symptoms, alone or in combination, that may produce a long-term functional disability (9). This definition also emphasized brain versus head injury, a distinction that has considerable potential implications for injury reporting, management and expectations of recovery. The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) includes postconcussive syndrome (PCS), which is characterized by headache, memory and attentional deficits, fatigue, irritability, depression, anxiety, sleep disturbance and apathy (10). In 1999, the DSM-IV and ACRM definitions were harmonized by Ruff and Jurica to subclassify mTBI into grades I, II and III (11). Type I is the original ACRM definition, type III the DSM-IV and type II bridges the gap between these two. In 2004, the second International Symposium on Concussion in Sport (ISCS) added two additional categories of concussion: simple, which resolves without complication in 7-10 days and does not require medical intervention, and complex, which includes persistent physical or cognitive symptoms with more severe effects including LOC greater than 1 min, convulsions, or other significant sequelae (12). The ISCS also recognized that repeated concussions could occur from progressively less impact force.
Clearly, the diversity with which mTBI and concussions are defined by medical professionals and recognized by individuals who have experienced mild head trauma raises great challenges in estimating their incidence, prevalence and consequences. A standardised approach to grade mTBI severity, ideally using a combination of neuropsychological, imaging and biochemical measures, will greatly improve the reliability and validity of clinical reports on the consequences of mTBI as well as drive the development of defined experimental model systems with which to investigate the potential mechanisms by which mTBI can produce long-term effects.
Sowing the seeds of dementia
An association between antecedent TBI and dementia risk is supported by the observation that some of the pathogenic mechanisms known to occur after brain injury involve proteins already known to contribute to the neuropathological hallmarks of AD, including extracellular amyloid plaques that are composed of aggregated β-amyloid (Aβ) peptides and intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein (13). Although the pathogenesis of AD is not completely understood, a leading hypothesis is that aberrant metabolism of Aβ peptides, derived from proteolytic cleavage of amyloid precursor protein (APP), triggers many of the toxic events in AD and eventually leads to both tau and amyloid deposition (14). Major support for this hypothesis has come from the updated Diagnostic Guidelines for Alzheimer’s Disease, released in April 2011, that include changes in cerebrospinal fluid (CSF) Aβ and tau levels as one of the earliest detectable events in the preclinical phase of AD, followed by the appearance of amyloid plaques, the loss of grey matter and ventricular enlargement, and finally by the onset of cognitive impairment (15).
Despite the involvement of Aβ and tau in the neuropathology of TBI and AD, the widespread white matter involvement in TBI that is not present in AD results in some noteworthy differences in the pattern and distribution of the common neuropathological features in the two entities. For example, the neuropathology of TBI is largely one of tau deposits, as approximately 30% of patients who suffer a severe TBI show Aβ deposits (16) that tend to be diffuse rather than mature amyloid plaques (21), suggesting a more dynamic phase of plaque development after TBI than in AD. In addition, diffuse Aβ plaques have been reported to appear hours after injury in short term survivors, but were observed to be largely absent in long term survivors of TBI where the mean survival time was 245 days (16). However, a very recent study examined survivors of a single moderate-severe TBI from 1-47 years after injury and observed widespread non-diffuse Aβ plaques (17), suggesting that a single TBI can induce long-term changes resembling AD neuropathology. In addition, this study was the first to report the appearance of NFTs after a single TBI compared to previous studies that had found NFTs in only mild, repetitive, concussive type injuries (17). Increased tau protein has also been found in the CSF of TBI patients and is correlated with the degree of axonal injury (18). Revisiting the prevalence of amyloid, Aβ and tau levels in CSF and brain tissue after TBI is likely to become an important avenue of investigation, particularly when factors such as age at injury, number of injuries, and severity of injuries are also considered.
Diffuse axonal injury, which occurs in over 50% of TBI cases (19), is induced by rapid acceleration or deceleration forces. APP accumulation in damaged axons is a striking histological hallmark of diffuse axonal injury (20;21). It has been hypothesized that increased APP in the post-TBI brain is accompanied by a burst of Aβ production, which can deposit in amyloid plaques similar to those found in the brains of AD patients (22-24). However, the effect of brain injury on Aβ dynamics is far from simple (25). Microdialysis experiments in human subjects shows that the levels of Aβ in interstitial fluid (ISF) correlates with the patient’s GCS, such that ISF Aβ levels increase as neurological status improves, remains unchanged in clinically stable patients, and declines as neurological status worsens (26;27). These remarkable observations suggest that Aβ release is associated with recovery of synaptic function, a conclusion supported by experiments in animal models (28). An important question now is how the presence of pre-existing amyloid deposits or APP-immunoreactive neurites may influence Aβ dynamics after brain injury. Microdialysis measurements of Aβ half-life in experimental animal models show that the presence of pre-existing amyloid plaques significantly slows the rate of Aβ decay (29). Additionally, APP-immunoreactive neuronal processes can be detected in both AD subjects and non-demented controls and are hypothesized to develop prior to neurofibrillary tangles (30). These observations suggest that brain injuries that occur in people old enough to have amyloid deposits or dystrophic neurites could exacerbate pathogenesis to a greater extent than in individuals without these markers at the time of injury.
Mild repetitive TBI is increasingly recognized to be associated with a neurodegenerative disease known as chronic traumatic encephalopathy (CTE) (24). CTE, which was originally identified in boxers and termed dementia pugilistica, presents with tremor, bradykinesia, confusion and speech impediments. CTE is also characterized by a progressive dementia and a pronounced accumulation of NFTs in the neocortex and brainstem. Approximately 30% of CTE cases also exhibit amyloid deposits, suggesting that CTE may be a distinct clinical entity from AD even though the same proteins likely contribute to its etiology. Progressive neurological deterioration has also been observed in other high contact sports including gridiron football, professional wrestling and ice hockey (24). A study of four mild chronic head injury cases found that NFT formation occurred in the absence of Aβ with cytoskeletal abnormalities accumulating around damaged blood vessels and perivascular elements (31). In experimental models, the formation of tau deposits can be uncoupled from amyloid deposits (32), suggesting that appearance of NFTs and Aβ deposits induced by brain injury can develop independently.
Epidemiological studies of TBI and dementia risk
The vast majority of epidemiological studies reporting on associations between TBI and dementia have used a retrospective case-control study design and many have low statistical power (Table 1, Figure 1). The highly-powered EURODEM meta-analysis pooled the findings of 11 case-control studies to investigate the association between head trauma and AD (33). This analysis reported a significant pooled relative risk of 1.82 (95% CI: 1.26-2.67) for head trauma associated with AD. Head trauma did not reduce the age of onset of AD, even when stratified by familial and sporadic AD. However, a trend was observed in the pooled data with respect to increased risk as the time between the last head trauma and AD onset diminished. Although not significant, the estimated relative risk for head trauma within 10 years of AD onset was 5.53, more than three-fold greater than the relative risk for head trauma occurring more than 10 years prior to onset, which was 1.63. This observation suggests that there may be a critical age range where head trauma increases dementia risk, as advancing age will eventually outweigh other factors that elevate AD risk.
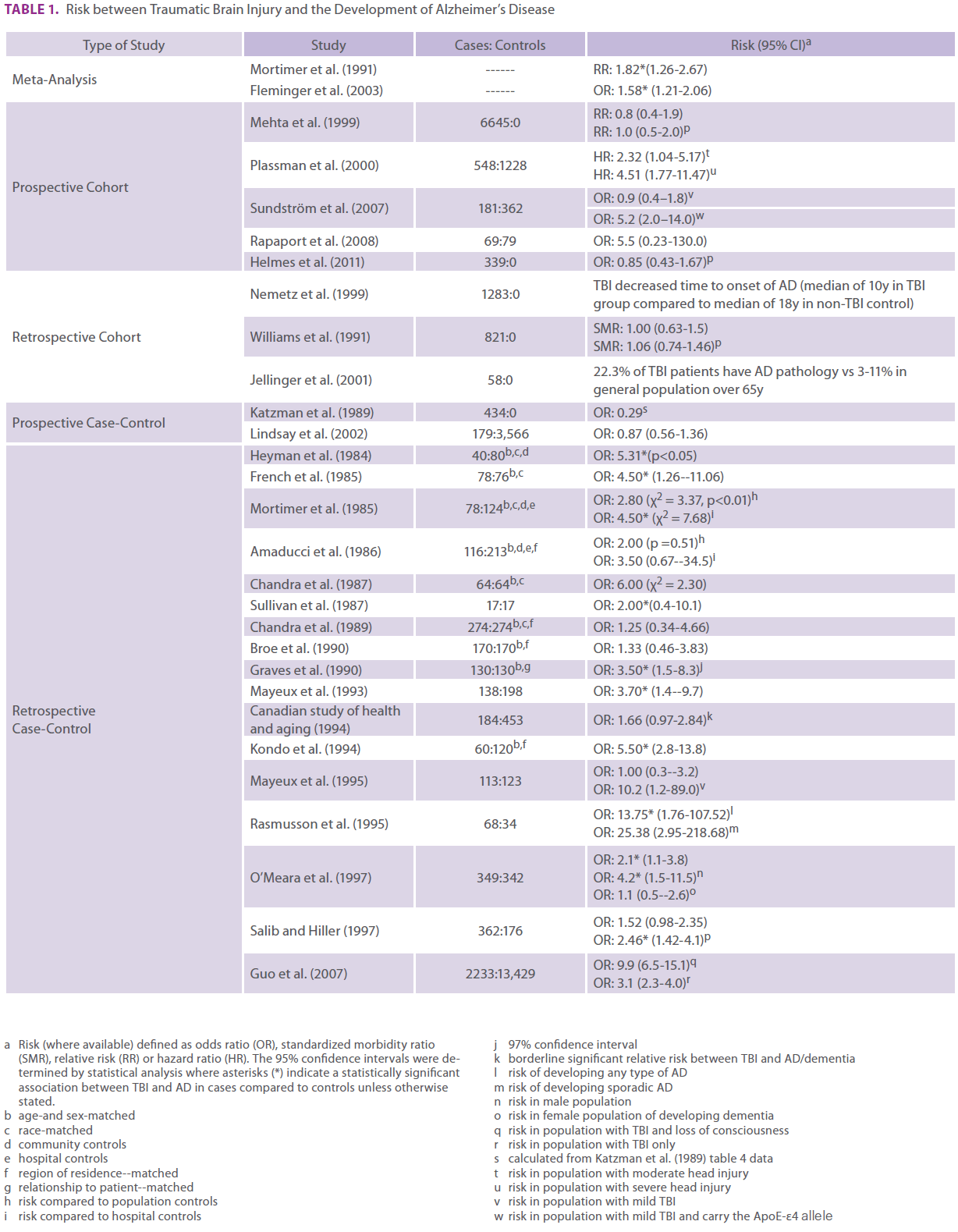
Table 1: Risk between Traumatic Brain Injury and the Development of Alzheimer’s Disease
Limitations of retrospective case control studies include their reliance on reports of previous head injuries by patients or their surrogates in the absence of documented evidence, which can lead to potential recall bias. Additionally, LOC associated with head injury may not represent an accurate proxy of injury severity. Another difficulty in identifying an association between TBI and AD is the lack of a neuropathologically confirmed AD diagnosis, potential leading to a high false-positive rate in many studies.
Prospective cohort studies are beginning to address some of these limitations. For example, the Nemetz et al. populationbased study was designed to test whether time to onset of AD is reduced after head injury (Table 1, Figure 1) (34). Data were collected from all documented episodes of brain injury from 1935-1984 among Omstead County, Minnesota, residents. Of the 1,283 head injury cases aged 40 and over who were followed, 31 developed AD (2.4%), which is within the standardized incidence ratio of 1.2 (95% CI: 0.8-1.7). However, subjects with antecedent head injury had a significantly (p=0.015) reduced time to onset of AD (median 10 years) than those without head injury (median 18 years), suggesting that head injury may reduce time to onset of AD. It should be noted that inclusion criteria for TBI in this study included LOC, post-traumatic amnesia or neurologic signs of brain injury, and that these subgroups were not considered separately when evaluating time to onset of AD. It is possible that the above subgroups represent different severities of TBI and may differ in how they influence time to onset of AD. When stratified by age at injury, the association was stronger for cases where injury occurred prior to age 65 (p<0.001), however, injuries after the age of 65 years did not reduce the time to AD onset (p=0.867). The authors conclude that head injury increased AD risk only if the injury occurred before age 65. One of the major strengths of this study is the documentation of head injuries rather than reliance on self- or proxy-reports
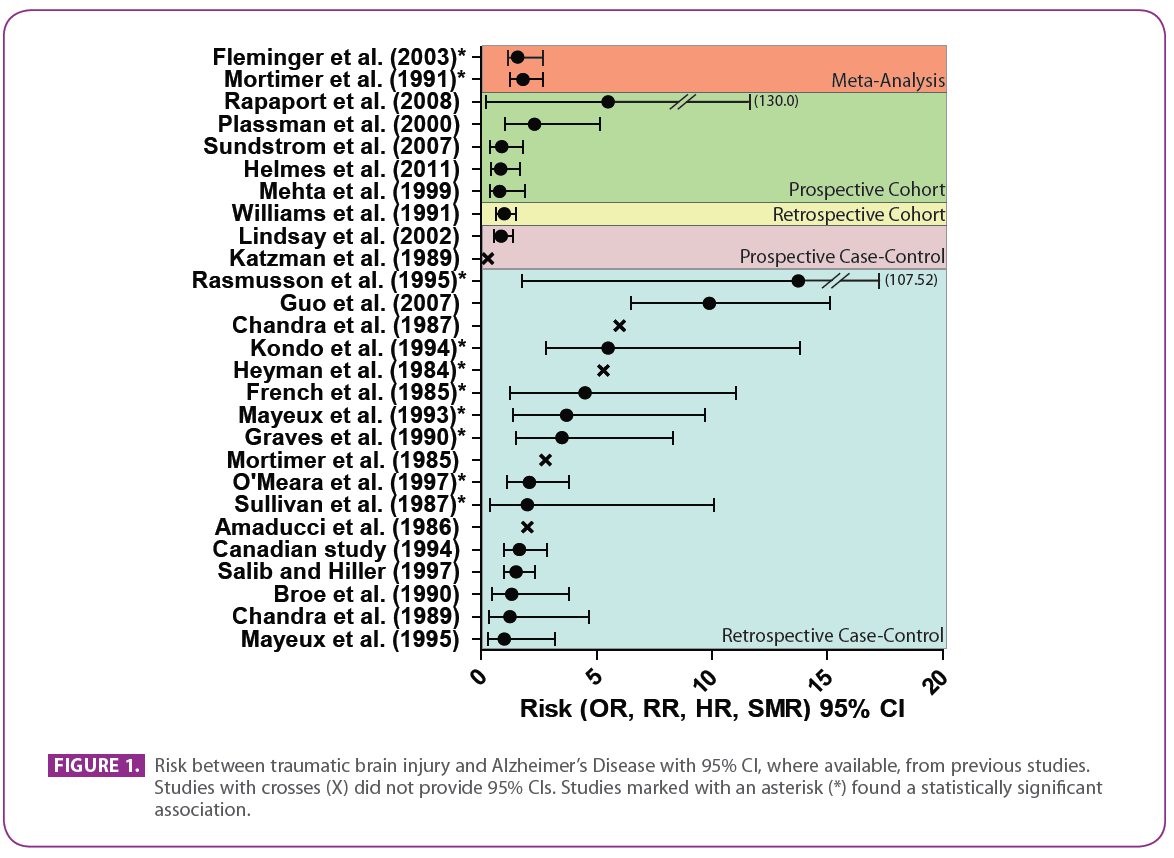
Figure 1: Risk between traumatic brain injury and Alzheimer’s Disease with 95% CI, where available, from previous studies. Studies with crosses (X) did not provide 95% CIs. Studies marked with an asterisk (*) found a statistically significant association.
Filling the knowledge gaps
A definitive answer to the question of whether antecedent TBI increases dementia risk will require careful attention to several components of study design. For example, a prospective cohort design with documentation on head injury frequency and severity is of utmost importance. Increasing public health knowledge about the potential long-term consequences of concussions may increase the frequency with which mTBI is reported and lead to a more representative sampling of brain injured subjects from the general population.
Although not routinely possible, inclusion of baseline data in high-risk populations is highly encouraged and could be obtained upon recruitment to elite sports teams or upon entry into military service. Pre-injury data will increase sensitivity of cohort studies because results will not rely on normative population data. In addition, since athletes represent a relatively homogenous population of young healthy individuals, studying this population may help somewhat to reduce the challenge of comorbidities often observed in subjects with sporadic brain injuries. The athletic population also offers considerable advantages to investigating associations between mTBI and dementia, largely because of the sheer number of people who participate in sports at some time in their lives. For example, soccer, or association football (AF), is the most popular sport worldwide with over 240 million currently active players (35). Studies on high-risk populations may therefore address some of the limitations of low statistical power in many of the existing reports.
Toward this goal, Barth has pioneered a baseline-serial testing paradigm delivered at 24h, 5d and 10d after concussion that detects residual symptoms in attention, working memory, verbal memory, visuospatial memory, verbal learning, information- processing speed, reaction time and executive functions (36). A meta-analysis of 21 studies involving 790 athletes and 2, 014 controls reported altered global functioning, memory acquisition and delayed memory performance 24h after injury (37). The vast majority of concussed subjects return to baseline neuropsychological functioning within 10d of injury (37), however, it is possible that some have residual effects that are below the detection limit of current neuropsychological scales and increase the risk of repeated injury after return to play. It is also possible that there are biochemical sequelae of concussion that are not currently assessed or factored into return to play decisions.
However, studying head injuries only in the athletic population is not without additional caveats that may limit generalization of the results to the normative population. The mechanical forces involved in sport-related concussions may be quite different from other types of TBI injuries. For example, TBI induced by motor vehicle accidents (MVA) often includes higher acceleration/deceleration forces and a higher incidence of peripheral injuries that may increase the risk of debilitating postinjury depression, anxiety and pain (3). Other risk factors for poor prognosis in MVA-induced TBI include substance abuse, poor premorbid cognitive ability, increased age and comorbid conditions such as hypertension. Differences in subject demographics and biomechanics may significantly contribute to distinct long-term outcomes following TBI in the sport-related compared to the non-sport-related settings.
Importantly, neuropsychological performance is influenced by motivation, a factor that may also limit the ability of neuropsychological data collected in athletes to be applied to the general population, particularly in the forensic setting. Because athletes are generally highly motivated to return to play, it is possible to observe improved neuropsychological performance post injury compared to baseline (38). Conversely, subjects motivated by potential financial gain after injury repeatedly score poorly on neuropsychological batteries (38). Ensuring that neuropsychological examinations are controlled for motivation and malingering is essential, and supplementing neuropsychological evaluations with imaging, genetic and biomarker data may be necessary to identify subjects still at risk even though their neuropsychological performance appears to have recovered completely (39). Long term cognitive decline following head injury may also be influenced by other genetic and demographic variables including cognitive reserve. For example, one study that examined Vietnamese War veterans who had suffered penetrating head injuries (PHI) found that pre-injury intelligence was the strongest predictor of the degree of cognitive decline experienced by sufferers of PHI (40).
Being non-invasive, imaging methods hold tremendous potential in helping to establish injury severity and predicting outcome. MRI approaches can identify neurological evidence of axonal injury after TBI including CSF changes, white matter changes, subtle brain volume loss, hemorrhagic lesions, changes in prefrontal cortex cerebral blood flow during work ing memory tasks, dilated periventricular space, white matter inflammation and hemosiderin deposits (41;42). In particular, methods such as susceptibility weighted imaging (SWI) that are highly sensitive to post-traumatic microhemorrhages may become particularly important diagnostic and prognostic tools (41;42). Methods to visualize Aβ or, if developed, tau deposits in the living human brain may also find considerable application for long-term post-TBI management (43).
Developing validated CSF or plasma biomarkers of TBI may offer additional insights into monitoring recovery after injury and assessing long-term risk (44). To this end, a study of 14 amateur Swedish boxers showed that CSF total tau, neurofilament light protein and glial fibrillary acidic protein were significantly elevated one week after injury and declined by three months post injury, with higher levels associated with increased number and severity of punches received (45). Importantly, the new diagnostic guidelines for AD include, for research purposes, biomarkers that may be predictive of AD progression and appear 10-20 years before a traditional clinical diagnosis is made (15). Importantly, these AD biomarkers were developed from subjects excluded for brain injury. It is now of enormous importance to understand how TBI may alter the trajectory of AD biomarkers and whether TBI may produce a distinct biomarker signature that differs from that observed in the typical lateonset AD population.
What we can learn
There are considerable challenges to deducing whether or not TBI increases dementia risk. With respect to subject population, these challenges include the broad spectrum of TBI injury severities and types and the enormous diversity in subject demographics, such as age, gender and co-morbid conditions may greatly affect TBI outcome and dementia risk. In addition to the caveats of case-control studies discussed above, major challenges of study design also include lack of TBI documentation and standardization of definitions especially for mild injury and lack of genetic, biomarker, imaging and post-mortem neuropathological analyses. It is possible that TBI may trigger a type of dementia that shares some neurological, pathological, imaging and molecular features as other dementias such as AD but may also include certain distinguishing features. TBI is likely to alter the natural history of conditions such as AD, and an important avenue for further research will be to determine how the multitude of variables associated with TBI may or may not affect the pathological and clinical progression toward dementia. Sufficiently powered studies that collect standardised data across neuropsychological, imaging, genetic and molecular domains will be required to untangle the skein of possible outcomes from brain injury.
2205
References
- Leon-Carrion J, Dominguez-Morales Mdel R, Barroso YMJM, Murillo-Cabezas F. Epidemiology of traumatic brain injury and subarachnoid hemorrhage. Pituitary 2005;8:197-202.
- Summers CR, Irving B, Schwab KA. Traumatic brain injury in the United States: an epidemiologic overview. Mt Sinai J Med 2009;76:105-10.
- Bailey CM, Barth JT, BEnder SD. SLAM on the stand: How the sports-related concussion literature can inform the expert witness. J Head Trauma Rehabil 2009;24:123-30.
- Grimm D, Mion LC. Falls resulting in traumatic injury among older adults: nursing care issues. AACN Adv Crit Care 2011;22:161- 8.
- Elder GA, Cristian A. Blast-related mild traumatic brain injury: mechanisms of injury and impact on clinical care. Mount Sinai J Med 2009;76:111-8.
- Chen AY, Colantonio A. Defininig neurotrauma in administrative data using the International Classification of Diseases Tenth Edition. Emerging Themes in Epidemiology 2011.
- Teasdale G, Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet 1974;304:81-4.
- Barth JT, Freeman JR, Broschek DK. Mild head injury. In: Ramachandran VS, editor. Encyclopedia of the human brain. Vol 3 ed. San Diego: Academic Press; 2002.
- Kay T, Harrington D, Adams R, Berrol S, Cicerone K, Dahlberg C, et al. Definition of mild traumatic brain injury. J Head Trauma Rehabil 1993;8:86-7.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4 ed. Washington, DC: 1994.
- Ruff RM, Jurica P. In search of a unified definition for mild traumatic brain injury. Brain Inj 1999;13:943-52.
- McCrory P, Johnston K, Meeuwisse W, Aubry M, Cantu R, Dvorak J, et al. Summary and agreement statement of the 2nd International Conference on Concussion in Sport, Prague 2004. Clin J Sport Med 2005;15:48-55.
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 2001;81:741-66.
- Yankner BA, Lu T. Amyloid beta-protein toxicity and the pathogenesis of Alzheimer’s disease. J Biol Chem 2009;284:4755- 9.
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging- Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s Disease. Alzheimer’s and Dementia 2011;7:280-92.
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid b plaques despite persistent accumulation of amyloid b in axons of long-term survivors of traumatic brain injury. Brain Pathol 2009;19:214-23.
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloidbeta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2011;epub.
- Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, et al. Quantification of axonal damage in traumatic brain injury: Affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem 1999;72:741-50.
- 19) Adams JH, Jennett B, Murray LS, Teasdale GM, Gennarelli TA, Graham DI. Neuropathological findings in disabled survivors of a head injury. J Neurotrauma 2011;28:701-9.
- Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J Neurotrauma 1995;12:565-72.
- McKenzie JE, Gentleman SM, Roberts GW, Graham DI, Royston MC. Increased numbers of beta APP-immunoreactive neurones in the entorhinal cortex after head injury. Neuroreport 1994;6:161-4.
- Horsburgh K, Cole GM, Yang F, Savage MJ, Greenberg BD, Gentleman SM, et al. beta-amyloid (Abeta)42(43), abeta42, abeta40 and apoE immunostaining of plaques in fatal head injury. Neuropathol Appl Neurobiol 2000;26:124-32.
- Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 1990;53:373-8.
- Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 2011;30:179-88.
- Magnomi S, Brody DL. New perspectives on amyloid-b dynamics after acute brain injury. Arch Neurol 2011;67:1068-73.
- Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialsis in patients with traumatic brain injury. J Neurosurg 2009;110:1227-37.
- Brody DL, Magnoni S, Schwetey KE, Spinner ML, Esparza TJ, Stocchetti N, et al. Amyloid-b dynamics correlate with neurological status in the injured human brain. Science 2008;321:1221-4.
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 2005;48:913-22.
- Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changed in amyloidbeta metabolism and half-life. J Neurosci 2003;23:8844-53.
- Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A 1991;88:7552- 6.
- Geddes JF, Vowles GH, Nicoll JA, Révész T. Neuronal cytoskeletal changes are an early consqeuence of repetitive head injury. Acta Neuropathol 1999;98:171-8.
- Tran HT, LaFerla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-b accumulation and independently accelerates the development of tau abnormalities. J Neurosci 2011;31:9513-25.
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20 Suppl 2:S28-S35.
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, et al. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol 1999;149:32-40.
- Rutherford A, Stephens R, Potter D. The neuropsychology of heading and head trauama in Association Football (Soccer): A review. Neuropsychol Rev 2003;13:153-79.
- Barth JT, Alves WM, Ryan TV. Mild head injury in sports: neuropsychological sequelae and recovery of function. In: Levin HS, Eisenberg HM, Benton AL, editors. Mild Head Injury.New York: Oxford University Press; 1989. p. 257-75.
- Belanger HG, Vanderploeg RD. The neuropsychological impact of sports-related concussion: a meta-analysis. J Int Neuropsychol Soc 2005;11:345-57.
- Bailey CM, Echemendia RJ, Arnett PA. The impact of motivation on neuropsychological performance in sports-related mild traumatic brain injury. J Int Neuropsychol Soc 2006;12:475-84.
- Ellenberg D, Henry LC, Macciocchi SN, Guskiewicz KM, Broglio SP. Advances in sport concussion assessment: from behavioral to brain imaging measures. J Neurotrauma 2009;26:2365-82.
- Raymont V, Greathouse A, Reding K, Lipsky R, Salazar A, Grafman J. Demographic, structural and genetic predictors of late cognitive decline after penetrating head injury. Brain 2008;131:543-58.
- Chastain CA, Oyoyo UE, Zipperman M, Joo E, Ashwal S, Shutter LA, et al. Predicting outcomes of traumatic brain injury by imaging modality and injury distribution. J Neurotrauma 2009;26:1183-96.
- Kim JJ, Gean AD. Imaging for the diagnosis and management of traumatic brain injury. Neurotherapeutics 2011;8:39-53.
- Weiner MW, Aisen PS, Jack CRJr, Jaqust WJ, Trojanowski JQ, Shaw L, et al. The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement 2010;6:202-11.
- Dash PK, Zhao J, Hergenroeder G, Moore AN. Biomarkers for the diagnosis, prognosis, and evaluation of treatment efficacy for traumatic brain injury. Neurotherapeutics 2010;7:100-14.
- Zetterberg H, Hietala MA, Jonsson M, Andreasen N, Styrud E, Karlsson I, et al. Neurochemical aftermath of boxing. Arch Neurol 2006;63:1277-80.

