Keywords
Calcium, Exocutosis, EGTA
Abbreviations
NE: norepinephrine, PKC: protein kinase C, CaM: calmodulin, SNAP-25: synaptosome-associated protein of 25 kDa, CAPS: calcium-dependent activator protein for secretion, SNARE, SNAP: (soluble N-ethylmaleimide-sensitive factor attachment proteins) receptor, CaMK: Ca2+/calmodulin-dependent protein kinase, PAGE: polyacrylamide gel electrophoresis, AMP-PNP: adenosine 59-(b,g- imido) triphosphate, HA: hydroxyapatite
Introduction
The molecular mechanisms of presynaptic vesicle release have been extensively examined by a combination of biochemical, genetic, and electrophysiological techniques. A series of proteinprotein interaction cascades have been proposed to lead to vesicle docking and fusion [1–3]. The SNARE protein family, including syntaxin, SNAP-25, and vesicle-associated membrane protein (VAMP, also called synaptobrevin), plays an essential role in promoting membrane fusion, andis thought to comprise the basic fusion machinery [4,5]. In Ca2+-stimulated exocytosis, many additional proteins are important in the Ca2+ regulation of the basic membrane trafficking apparatus. Calcium not only triggers rapid fusion of release-competent vesicles, but is also involved in earlier processes which replenish the pool of readily releasable vesicles [6]. Furthermore, it appears to be critical in initiating several forms of synaptic plasticity including posttetanic potentiation [7]. The molecular mechanisms by which Ca2+ regulates these processes is not well understood. PC12 cells have often been utilized to study Ca2+-activated exocytosis, as they offer a homogeneous cell population that possesses the same basic exocytic machinery as neurons [8].
In this study, we used an established cracked cell assay, in which [3H]norepinephrine (NE)1 labeled PC12 cells are permeabilized by mechanical “cracking” and then reconstituted for secretion of NE in the presence of test proteins [9]. Transmitter-filled vesicles and intracellular cytoskeletal structures remain intact in these cells, while cytosolic proteins leak out [10]. These cracked cells readily release NE upon addition of ATP, brain cytosol, and 1 mM free Ca2+ at an elevated temperature.
We term this a “composite assay,” as all essential components are added into one reaction mixture. Alternatively, cracked cells can be first primed with cytosol and ATP, washed, then reconstituted for NE release with cytosol and Ca2+ [11]. This sequential priming-triggering protocol is useful for determining whether a protein acts early or late in the exocytic pathway, and whether its effect is dependent on Ca2+ or ATP. This semiintact cell system serves as a bridge between an in vitro system comprised of purified components, and electro-physiological systems that monitor release in vivo. It provides information on protein functions in a cell with an intact membrane infrastructure while being easily manipulatable. Ca2+ regulation by membrane depolarization is no longer a concern, as intra-cellular Ca2+ concentration can be controlled by a buffered solution. Indirect readout of neurotransmitter release using a postsynaptic cell is replaced by direct readout of [3H]NE released into the buffer. Complications associated with interpreting overlapping exo- and endocytotic signals are also eliminated as only one round of exocytosis is measured.
Finally, concentration estimates are likely to be accurate, since added compounds do not need to diffuse long distances along axons and dendrites to their sites of action. Using this assay, several proteins required for NE release have been purified from rat brain cytosol, including phosphatidyl-inositol transfer protein, phosphatidylinositol-4-phosphate 5-kinase [12], and calcium-dependent activator protein for secretion (CAPS) [9]. The validity of the cracked cell system is confirmed by the finding that phosphatidylinositol transfer protein and CAPS are mammalian homologues of yeast SEC14p [13] and nematode UNC31p, respectively [14], both proteins involved in membrane trafficking [15, 16].
Calmodulin is the most ubiquitous calcium mediator in eukaryotic cells, yet its involvement in membrane trafficking has not been well established. Some early studies showed that calmodulin inhibitors [17–19], anti-calmodulin antibodies [20,21], or calmodulin binding inhibitory peptides [22] inhibited Ca2+- activated exocytosis. However, in other studies, calmodulinbinding peptides and an anti-calmodulin antibody led to the conclusion that calmodulin is only involved in endocytosis, not exocytosis [23]. More recently, it was reported that Ca2+/ calmodulin signals the completion of docking and triggers a late step of homotypic vacuole fusion in yeast, thus suggesting an essential role for Ca2+/calmodulin in constitutive intracellular membrane fusion [24]. If calmodulin indeed plays an important role in exocytosis, a likely target of calmodulin is Ca2+/calmodulindependent protein kinase II (CaMKII), a multifunctional kinase that is found on synaptic vesicles [25] and has been shown to potentiate neurotransmitter release [26,27].
Another Ca2+ signaling molecule, PKC, has also been implicated in regulated exocytosis. In various cell systems, it has been shown that the phorbol esters stimulate secretion [28,29]. It is usually assumed that phorbol esters effect on exocytosis is through activation of PKC, but Munc13- 1 was recently shown to be a presynaptic phorbol ester receptor that enhances neurotransmitter release [30,31], which complicates the interpretation of some earlier reports. The mode of action of PKC remains controversial. There is evidence that PKC increases the intracellular Ca2+ levels by modulating plasma membrane Ca2+ channels [32, 33], that it increases the size of the release competent vesicle pool [34], or that it increases the Ca2+ sensitivity of the membrane trafficking apparatus no consensus on these issues has been reached.
PKC substrates that have been implicated in exocytosis include
1. SNAP-25,
2. synaptotagmin [28],
3. CAPS, and
4. nsec1.
It is believed that upon phosphorylation, these PKC substrates might interact differently with their binding partners, which, in turn, leads to the enhancement of exocytosis. In addition, evidence is accumulating that PKC and calmodulin interfere with each others actions, as PKC phosphorylation sites are embedded in the calmodulin-binding domains of substrates such as neuromodulin and neurogranin. It is therefore possible that PKC could modulate exocytosis via a calmodulin-dependent pathway by synchronously releasing calmodulin from storage proteins.
In this study, we fractionated an EGTA extract of brain membranes in order to identify active components that could reconstitute release in the cracked cellassay system. We identified calmodulin and PKC as two active factors. Thus, we demonstrate that calmodulin and PKC play a role in the Ca2+ regulation of exocytosis, and provide further insight into the mechanisms of their action.
Experimental Procedures
Materials
Rat Brain Cytosol Preparation Membrane EGTA Extract Preparation
Cracked Cell Assay
PC12 cells were maintained and [3H]NE labeled as described previously [11]. Labeled cells were harvested by pipetting with ice-cold potassium glutamate buffer (50 mM Hepes, pH 7.2, 105 mM potassium glutamate, 20 mM potassium acetate, 2 mM EGTA) containing 0.1% bovine serum albumin. Subsequent manipulations were carried out at 0–4°C. Labeled cells (1–1.5 ml/dish) were mechanically permeabilized passage through a stainless steel homogenizer. The cracked cells were adjusted to 11 mM EGTA and incubated on ice for 0.5–3 h, followed by three washes in which the cells were centrifuged at 800 3 g for 5 min and resuspended in potassium glutamate buffer containing 0.1% bovine serum albumin.
Composite Assay
Each release reaction contains 0.5–1 million cracked cells, 1.5 mM free Ca2+, 2 mM MgATP, and the protein solution to be tested in potassium glutamate buffer. Release reactions were initiated by incubation at 30°C and terminated by returning to ice. The supernatant of each reaction was isolated by centrifugation at 2,500 3 g for 30 min at 4°C, and the released [3H]NE was quantified by scintillation counting (Beckman LS6000IC). Cell pellets were dissolved in 1% Triton X-100, 0.02% azide and similarly counted. NE release was calculated as a percentage of total [3H] in the supernatant.
Priming Assay
A priming reaction contains about 1–2 million cracked cells, 2 mM MgATP, and the protein solution to be tested. Ca2+ is omitted.
The primed cells were spun down, washed once with fresh potassium glutamate buffer, and distributed into two triggering reactions, each containing rat brain cytosol and free Ca2+. The triggering reaction was performed at 30°C for 3 min, and the NE release was measured as in a composite assay.
Triggering Assay
Cracked cells were primed, centrifuged, washed, and distributed into triggering reactions containing 1.5 mM free Ca2+ and the protein solution. To inhibit any ATP dependent activity in the triggering reaction, an ATP depletion system of hexokinase, MgCl2, glucose or a non-hydrolyzable ATP analogue AMPPNP was added into the triggering reaction. NE release was measured as above.
Free Ca2+ Concentration Determination
The range of Ca2+ free in the release reaction (Figure 1) was achieved by adding Ca2+ into potassium glutamate buffer to reach final [Ca2+] total values of 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 1.9, and 2.0 mM. The pH of the reaction was 7.24 when no Ca2+ was added and 7.04 when 2.0 mM Ca2+ was added, in the absence of protein extracts or cracked cells.

Figure 1: Cytosol prepared in the absence of EGTA.
Free Ca2+ concentrations were determined using video microscopic measurements of fura-2 fluorescence. [Ca2+] free was calculated from the equation [Ca2+] free 5 Kd*3 (R 2 Rmin)/(Rmax 2 R).
The values of Rmin, Rmax, and Kd* were determined in the following solutions:
Potassium glutamate buffer (PGB) containing 8 x 3 10^6 cracked cells/ml, 2 mM MgATP (PGB+CC)
1) Rmin: PGB+CC and 10 mM additional EGTA,
2) Rmax: PBG+CC, and 10 mM total Ca2+,
3) Kd*: PGB+CC, 28 mM additional EGTA, and 18 mM total Ca2+, pH 7.2 ([Ca2+]free 5 = 169 nM, determined in the absence of cells and MgATP based on fura-2 calibration in cell-free solutions).
These solutions were incubated at 37°C, mixed with fura- 2 pentapotassium salt (100 mM, Molecular Probes, Eugene, OR), and imaged. This procedure allowed us to take into account changes in fura-2 properties caused by the presence of permeabilized cells. Duplicate measurements of the above range of [Ca2+] total gave the following average [Ca2+] free values: 106, 146, 277, 462, 971, 1468, 1847, and 2484 nM.
Purification of Active Proteins
All procedures were carried out at 4°C or on ice. Membrane EGTA extract of one or two bovine brain(s) was filtered through cheesecloth and loaded overnight onto a column packed with DEAE-Sepharose CL-6B beads (Amersham Pharmacia Biotech).
The column was then washed with (20 mM Hepes, pH 7.5, 0.25 mM sucrose, 2 mM EGTA, 1 mM dithiothreitol) and step eluted with 10 column volumes of elution buffer (20 mM Hepes, pH 7.5, 2 mM EGTA, 400 mM KCl, 1 mM dithiothreitol). 100 ml of every other fraction was dialyzed overnight into PGB, and tested in a composite release assay for activity. The active fractions were pooled and dialyzed into zero salt buffer (20 mM Hepes, pH 7.5, 2 mM EGTA) and batch bound to 10 ml of Affi-Gel Blue beads (Bio-Rad) or DyeMatrex-Green A beads (Amicon) Blue beads were used in earlier experiments, and Green beads were used later to specifically deplete CAPS, which was known to bind to Green beads [9].
The unbound material was collected, concentrated to about 2 ml using a Centriprep-10 (Amicon), and loaded onto a 120 ml HiPrep Sephacryl S-200 gel filtration column (Amersham Pharmacia Biotech).
Samples were run on the S-200 column in PGB at a flow rate of 7 ml/h. 10–50 ml of every other fraction was tested for activity in the cracked cell composite assay, and two peaks of activity were observed (Figure 2).

Figure 2: The EGTA extract of brain membranes can support NE release in the absence of cytosol. Rat brain membrane EGTA extract (closed triangles) and rat brain cytosol (closed squares) were prepared as described under “Experimental Procedures.” NE release was measured in a composite reaction mixture of cracked cells, MgATP, Ca2+, and the indicated amount of crude extracts.
The first peak of activity had a predicted molecular mass of 85 kDa. The corresponding material was adjusted to 10 mM potassium phosphate concentration (pH 7.2) and loaded onto a 1-ml column packed with hydroxyapatite Bio-Gel HT (Bio-Rad).
The bound material was eluted with a linear K-PO4 gradient from 10 to 500 mM (pH 7.2) at a flow rate of about 0.1 ml/min, and 0.4– 0.5-ml fractions were collected. Each fraction was dialyzed into PGB and tested for activity.
The fractions were also analyzed by SDS-PAGE and silver staining (Sigma silver stain kit). The active material was concentrate ed and resolved on an 8% poly-acrylamide gel.
Two Coomassie-stained protein bands that matched the activity profile (Figure 3) were excised from the gel, sequenced by the Stanford PAN facility.

Figure 3: Gel filtration chromatography reveals two stimulatory factors in the membrane EGTA extract.
The two polypeptide sequences obtained from the upper band were:
1. LLNQEEGEYYNVPIXEGD
2. IRSTLNPRWDESFT.
The only bovine protein that contains both polypeptides is PKCa.
The four polypeptide sequences obtained from the lower band were:
1. YELTGKFERLIVGLMRPPAY
2. LIEILASRTNEQIHQLVAA
3. MLVVLLQGTREEDDVVSEDL
4. EMSGDVRDVFVAIVQSVK.
Based on these sequences, the protein band was unambiguously identified to be bovine annexin VI. The second S-200 peak has a predicted molecular mass of 25 kDa. The corresponding material was dialyzed into zero salt buffer (20 mM Tris, pH 7.5, 1 mM EGTA) and injected onto a Mono-Q HR 5/5 FPLC column (Pharmacia). The FPLC run was performed at 18°C at 1 ml/min and 1-ml fractions were collected with a linear salt gradient from 0 to 1 M KCl over 71 ml. The fractions containing proteins (determined by A280) were dialyzed into PGB and tested in the cracked cell assay.
Western Blot
Anti-calmodulin antibody and anti-PKC antibody were used, and ECL (Amersham) was used for detection.
Results and Discussion
A Membrane EGTA Extract Supports NE Release. Brain cytosol, prepared as the supernatant of the brain homogenate, effectively stimulates NE release, in the cracked cell assay (Figure 4) as pr eviously shown [9].

Figure 4: The low molecular wen.ight active factor is calmodulin.
A. The membrane EGTA extract from one bovine brain (Start) was subjected to sequential fractionation on DEAE, Blue A, and Sephacryl S-200 columns. The pooled material containing the activity after each chromotographic step was analyzed by SDS-PAGE and Coomassie staining. The arrowheads indicate the presence of calmodulin in all the lanes. Calmodulin shows a mobility shift depending on whether or not Ca2+ is present during electrophoresis (see panel C).
B. The active material pooled from Sephacryl S-200 was fractionated on a Mono-Q FPLC column and the fractions (5 ml/fraction) were tested for activity in a composite assay. The activity peak is shown.
C, the active Mono-Q fractions (5 ml/fraction) were subjected to SDS-PAGE in the presence of 1 mM EGTA or 0.1 mM Ca2+, and the gels stained with Coomassie Blue.
D, fraction 47 (1 ml) was probed by Western blotting with a monoclonal anti-calmodulin antibody. No Ca2+ or EGTA was added during SDS-PAGE.
We wondered whether crude extracts other than cytosol could support NE release, and we focused on extractable peripheral membrane proteins.
We found that a salt or EGTA extract of brain membranes, membranes defined as the 100,000 3 g pellet of the crude homogenate, reconstituted secretion in the absence of cytosol. The salt extract only slightly enhanced NE release above background (data not shown), the EGTA extract not only stimulated NE release to a high level, similar to that supported by cytosol, but also had a higher specific activity than cytosol (Figure 4).
The ability of the membrane EGTA extract to support secretion is consistent with the fact that following cracking, the cells are immediately extracted with EGTA, and are presumably devoid of most membrane EGTA-extractable factors.
This also suggests that these factors, some of which are probably Ca2+-dependent membrane-associating proteins, participate in Ca2+- triggered exocytosis.
The Membrane EGTA Extract Is Enriched in Triggering Fators NE release in cracked cells can be resolved into two sequential stages, proteins can be tested for activity in either stage.
• an ATP-dependent priming stage
• an ATP-independent Ca21-dependent triggering stage [11]
An effect in priming indicates an early role for the protein, an effect in triggering a late ATP-independent role. Since the protein composition of the membrane EGTA extract and cytosol are different, we tested whether they had different activities in the priming stage versus the triggering stage.
We found that the membrane EGTA extract is enriched in factors that act during triggering stage of NE release, as the same amount of protein from the membrane EGTA extract as cytosol gave a higher stimulation in the triggering assay, but not in the priming assay (Figure 1).
Regular cytosol is prepared in a buffer containing 2 mM EGTA, and thus presumably contains some of the proteins present in the membrane EGTA extract.
Cytosol prepared in the absence of EGTA showed an even lower specific activity in the triggering assay compared with regular cytosol (Figure 1).
Identification of Calmodulin as an Active Triggering Factor in the EGTA Extract Biochemical fractionation of the bovine brain membrane EGTA extract was carried out to identify the active components capable of reconstituting NE release.
Activity was assayed in a composite reaction mixture containing
• cracked cells
• ATP
• Ca2+
• the test protein(s).
Except for the presence of bovine serum albumin in the basal buffer,no other proteins were added to the cell ghosts except for the test protein(s).
Initial tests indicated that at least part of the activity in the membrane EGTA extract binds to and can be efficiently eluted from an anion exchanger and hydroxyapatite resin, but does not bind to Amicon color resins.
The starting material was, therefore, sequentially purified using DEAE, Affi-Gel Blue (or Matrex Green-A), and gel filtration chromotography.
Gel filtration fractionation indicated the presence of two peaks of activity with predicted molecular masses of 25 and 85 kDa, respectively (Figure 3).
In order to purify the active component(s) in the membrane EGTA extract, the crude extract from one bovine brain was fractionated chromatographically (see Experimental Procedures” for details). Fractions from a Sephacryl S-200 gel filtration column were tested for their activity in stimulating NE release in the composite assay. The two activity peaks have predicted molecular masses of 85 and 25 kDa, respectively. The arrows indicate the retention volume of standard proteins run on the same column.
The low molecular weight active factor was purified to homogeneity, as judged by a Coomassie-stained SDS-PAGE gel, after a subsequent Mono-Q fractionation (Figure 4).
We reasoned that the protein might be calmodulin (43) based on the following:
1) It is a relatively small protein (14–18 kDa) that is abundant in the starting extract (Figure 4A). 2) It elutes at a very high salt concentration (0.41 M KCl) on the Mono-Q column. 3) It stains negatively in silver stain (data not shown) (Figure 4B). 4) Its electrophoretic mobility shifts depending on the presence or absence of Ca2+ (Figure 4C).
A Western blot with an anti-calmodulin monoclonal antibody gave a positive signal (Figure 4D), confirming our prediction.
Properties of Calmodulin-stimulated Exocytosis
We used commercial calmodulin or bacterially expressed recombinant calmodulin to confirm our purification result, both sources of authentic calmodulin stimulated NE release as expected. Moreover, we found that calmodulin stimulates secretion in a triggering assay as well as in a composite assay (Figure 5A).

Figure 5: Calmodulin stimulates NE release in the triggering stage.
A. calmodulin (obtained from Sigma) increased NE release in the triggering assay in a dosedependent fashion, in the absence of ATP or any other cytosolic proteins. In this particular experiment, the maximal release achieved by addition of rat brain cytosol was 46.5%.
B. the triggering assay was performed with different concentrations of free Ca2+. Calmodulin (3 mg bacterially expressed recombinant protein, closed squares) increased NE release with a similar Ca2+
sensitivity to rat brain membrane EGTA extract (10 mg, closed triangles), as compared with conditions in which no protein was added (open squares).
The half-maximal increase was at 75 nM (250 ng/200 ml) final calmodulin concentration. This is within the broad range of affinities between calmodulin and its various targets and suggests that the interaction between calmodulin and its target molecule in exocytosis is in the physiological range. When the triggering reaction was performed at different Ca2+ concentrations, calmodulin increased NE release only at high [Ca2+] (0.4 – 2 mM) similar to the crude EGTA extract (Figure 5B), suggesting that calmodulin contributes to the triggering activity of the membrane EGTA extract. Calmodulin’s affinity for Ca2+ has been reported to be around 1 mM [25], consistent with the Ca2+ requirement for calmodulin-stimulated secretion that we observed.
Western analysis with commercial protein as standards indicated that calmodulin constitutes about 5% of total proteins in the rat brain membrane EGTA extract and about 2% of total proteins in the rat brain cytosol (data not shown).
In addition, a significant amount of calmodulin appears to be left in the washed cell ghosts (data not shown). Based on the activity of saturating levels of pure calmodulin (releasing 6–10% of total [3H]NE) and crude EGTA extract (releasing ,45% of total [3H]NE), we estimated that calmodulin accounts for 13–22% of total activity of the extract. Consistent with this, a high affinity calmodulin-binding peptide (CaMKIIa(291–312) (44), used at 5 mM) and an anti-calmodulin antibody (2 mg/200 ml) inhibited about 20% of the membrane EGTA extract-stimulated release (6.7 mg of extract added, data not shown).
We showed that calmodulin increased NE release in the triggering stage. Since regular triggering reactions were performed in the absence of any added ATP, this suggests that calmodulin enhanced secretion in an ATP-independent fashion. Furthermore, residual ATP in the cell ghosts did not play a role, since addition of a hexokinase ATP depletion system that can deplete millimolar concentrations of ATP within a few minutes [11] had little effect, as did addition of 5 mM AMPPNP, which blocks ATP-dependent enzymatic activity (Figure 6A).
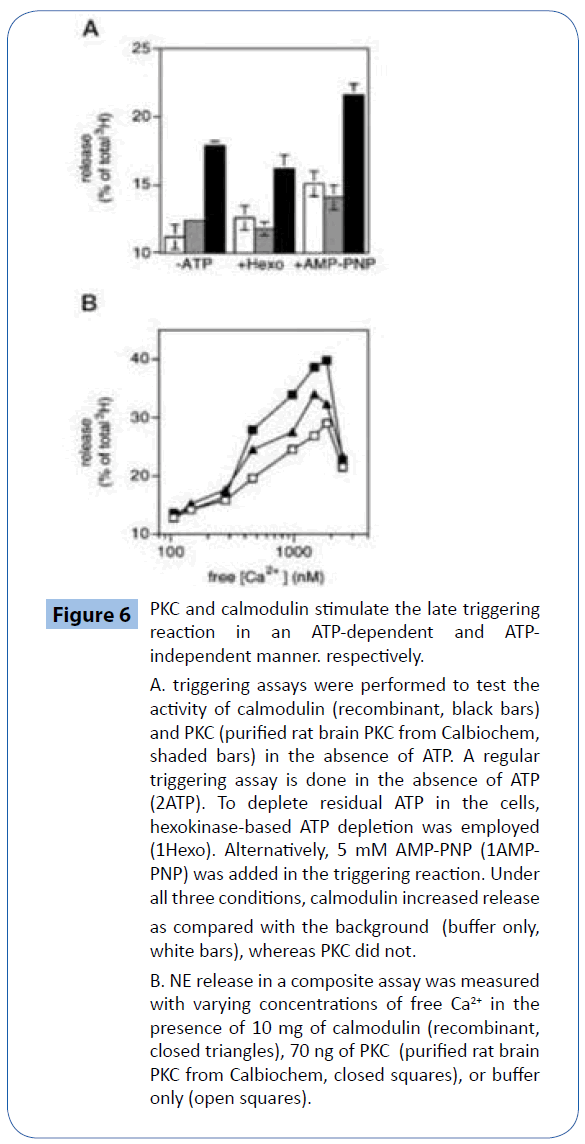
Figure 6: PKC and calmodulin stimulate the late triggering reaction in an ATP-dependent and ATPindependent manner. respectively.
A. triggering assays were performed to test the activity of calmodulin (recombinant, black bars) and PKC (purified rat brain PKC from Calbiochem, shaded bars) in the absence of ATP. A regular triggering assay is done in the absence of ATP (2ATP). To deplete residual ATP in the cells, hexokinase-based ATP depletion was employed (1Hexo). Alternatively, 5 mM AMP-PNP (1AMPPNP) was added in the triggering reaction. Under all three conditions, calmodulin increased release as compared with the background (buffer only, white bars), whereas PKC did not.
B. NE release in a composite assay was measured with varying concentrations of free Ca2+ in the presence of 10 mg of calmodulin (recombinant, closed triangles), 70 ng of PKC (purified rat brain PKC from Calbiochem, closed squares), or buffer only (open squares).
Therefore, we ruled out the possibility that a kinase mediates calmodulin’s effect.
A series of calmodulin mutants from Paramecium and chicken were tested for their ability to enhance Ca2+- stimulated secretion, and none of the mutations abolished the calmodulin effect (data not shown).
These mutations include S101F, M145V, E54K, G40E/D50N, V35I/D50N within Paramecium and calmodulin (45), and M124Q, M51A/V55A, and M51A/V55A/L32A within chicken calmodulin.
The Paramecium calmodulin mutants are the result of naturally occurring mutations that result in aberrations in their behavior. These mutants can be grouped into two categories according to their behavior, reflecting their loss of either a Ca2+-dependent Na1 current (calmodulin N-terminal lobe mutants: E54K, G40E/D50N, and V35I/D50N) or a Ca2+-dependent K1 current (calmodulin C-terminal lobe mutants: S101F and M145V).
The chicken calmodulin mutants have been shown to differentially activate myosin light chain kinase (M124Q, M51A/V55A, and M51A/V55A/L32A), CaMKII (M124Q), and CaMKIV (M124Q), and the mutated residues are thought to be important in defining calmodulin’s binding specificity.
Our finding that these mutant calmodulins can stimulate exocytosissuggests that calmodulin-binding domains similar to those of Paramecium Ca2+/calmodulin-dependent ion channels, myosin light chain kinase, CaMKII, and CaMKIV, are unlikely to mediate release utilizing the conserved SNARE fusion machinery, as they could be completely abolished by addition of exogenous syntaxin H3 domains (data not shown). the same molecular pathway was not activated, since their effects were additive (data not shown).
Discussion
In this study, we first identified an EGTA extract of brain membranes as a protein source capable of reconstituting Ca2+- activated exocytosis in cracked PC12 cells. EGTA only extracts a small pool of Ca2+-dependent membrane-associating proteins, it served as an efficient initial purification step.
Further protein chromatography led to the identification of two active factors in the starting extract, calmodulin and PKC, which together accounted for about half of the starting activity. Upon confirmation with commercially obtained proteins, this result unambiguously demonstrated that calmodulin and PKC mediate aspects of Ca2+-dependent processes in exocytosis.
The finding that brain membrane EGTA extract alone is able to replace cytosol in supporting Ca2+-triggered NE secretion in PC12 cells is somewhat surprising. We suggest that the likely explanation is 2-fold.
1. some cytosolic proteins essential for exocytosis have a membrane-bound pool within permeabilized cells, whose activity might be sufficient for a normal level of exocytosis.
2. Although the 100,000 3 g membrane pellet was washed to remove as many cytosolic proteins as possible, some cytosolic proteins that associate with membranes in a Ca2+-independent manner are probably present in the membrane EGTA extract.
These proteins likely constitute only a small percentage of the proteins in the extract, as the characteristics of the activity triggered by the membrane extract are quite different to that of cytosol (Figure 1).
Using an unbiased biochemical purification method, we demonstrated that calmodulin and PKC directly modulate the exocytotic machinery downstream of Ca2+ entry they signal through membrane-attached molecules to increase exocytosis.
These targets include integral and peripheral membrane proteins, and cytosolic proteins that have a significant membrane-bound pool. The modest stimulation by calmodulin and PKC on secretion might suggest a regulatory role. However, it is also possible that some intermediates in their signaling pathways are in limiting amounts in the cell ghosts, so that their full effects were not observed. Half-maximal stimulation was obtained at about 3 nM for PKC and at about 75 nM for calmodulin.
This is consistent with an enzymatic role for PKC, and predicts a high-affinity interaction between calmodulin and its substrate protein. Ca2+ regulates exocytosis at many different levels. Prior studies indicated that Ca2+ signaling occurs in the priming steps as well as in triggering steps.
Our priming triggering protocol does not allow Ca2+-dependent priming events to be assayed, as EGTA is present in the priming reaction. a different approach revealed the existence of both high and low Ca2+-dependent processes (Figure 1). this analysis indicated that late triggering events require high [Ca2+], whereas early priming events require low [Ca2+]. If, as proposed, there is a pronounced intracellular spatial and temporal [Ca] gradient from the point of Ca2+ entry during depolarization, perhaps triggered events occur closer to the point of Ca2+ entry, while Ca2+- dependent priming events occur further away from the point of Ca2+ entry. Distinct Ca2+ sensors at these stages might be appropriately tuned to different [Ca2+] to handle different tasks.
By analyzing the Ca2+ sensitivity of calmodulin-and PKC-stimulated release, we addressed the question of whether calmodulin and PKC plays an early or a late role in vesicle release. they both require relatively high [Ca2+] (Figure 6B), implying that calmodulin and PKC both mediate late triggering events, consistent with some earlier reports [34,35].
In addition, it is interesting to note that PKC does not alter the calcium sensitivity of release in cracked cells, in contrast to observations from the chick ciliary ganglion [36]. Therefore, in contrast to previous electrophysiological studies [28], we are able to limit the possible modes of PKC action in our system to an increase in the readily releasable vesicle pool or release sites, or an enhancement of the probability of release of individual vesicles upon Ca2+ influx.
The experiments assaying the calcium sensitivity of release (Figure 1, 5, and 6) demonstrated a drop in release at very high [Ca2+]. This decline in release at high [Ca2+] has been previously reported, and may represent the true Ca2+ sensitivity of the Ca2+-sensing mechanism inside cells. However, in our system, it could also be due to the activation of a variety of Ca2+ -activated proteases, as experiments are usually performed in the presence of crude extracts, which include unsequestered proteases. What might the molecular targets of PKC and calmodulin be? An obvious calmodulin target molecule is CaMKII. but calmodulin’s effect on exocytosis is ATP-independent, rendering the involvement of a kinase unlikely. Calmodulin has also been shown to associate with synaptic vesicles in a Ca2+-dependent fashion through synaptotagmin, probably by binding to its C-terminal tail, and to promote Rab3A dissociation from synaptic vesicles.
However, there was little calcium-dependent binding of calmodulin to synaptotagmin either on synaptic vesicles, in a bead binding assay with recombinant proteins, or in a calmodulin overlay (data not shown). In addition, using immobilized calmodulin, we did not see significant Ca2+- dependent pull-down of synaptotagmin or Rab3A from rat brain extract (data not shown).
Recent work has suggested three other candidate targets for calmodulin, Munc13, Pollux, and CRAG. Pollux has similarity to a portion of a yeast Rab GTPase-activating protein, while CRAG is related to Rab3 GTPase exchange proteins. Further work is required to investigate the role of their interactions with calmodulin in vivo. The recent report that calmodulin mediates yeast vacuole fusion [24] is intriguing, as it raises the possibility that calmodulin, a highly conserved ubiquitous molecule, may mediate many membrane trafficking events. It is not yet known if the effector molecule of calmodulin is conserved or variable across species and different trafficking steps.It is enticing to propose a model for Ca2+ sensing whereby.
calmodulin is a high affinity Ca2+ sensor for both constitutive and regulated membrane fusion.
1. In the case of constitutive fusion, calmodulin may be the predominant Ca2+ sensor.
2. In the case of slow, non-local exocytosis of large dense core granules, an additional requirement for
3. the concerted actions of other molecule(s) that are better tuned to intermediate rises in [Ca2+] might exist.
At the highly localized sites of fast exocytosis of small clear vesicles where high [Ca2+] is reached, specialized low affinity sensor(s) are likely required in addition to calmodulin to achieve membrane fusion.
Therefore, although calmodulin participates in multiple types of vesicle fusion, the impact of Ca2+ sensing by calmodulin on vesicle release likely varies. Due to the fact that calmodulin binding to some proteins can be modulated by PKC phosphorylation, one might suspect PKC action on exocytosis proceeds through a calmodulin-dependent pathway but the effects of calmodulin and PKC are additive within our system, suggesting that PKC does not act by releasing calmodulin from a substrate that functions as a calmodulin storage protein. How Ca2+ regulates presynaptic vesicle release has been an open question for many years. By identifying calmodulin and PKC as modulators of Ca2+-regulated exocytosis and clarifying their functions, we have extended our knowledge of the release process. While the basic machinery of membrane fusion is becoming better understood, the multiple effects of Ca2+ on exocytosis remain to be elucidated at the molecular level. In addition, the ways that Ca2+ regulation may be important to the mechanisms of synaptic plasticity in the central nervous system
Acknowledgment
We thank Diana Bautista and Dr. Richard S.Lewis for generous help with [Ca2+] free determination, Dr. Ching Kung for providing the Paramecium calmodulin mutants, and Dr. Anthony R. Means for providing the chicken calmodulin mutants. We also thank Dr. Jesse C. Hay for the initial setup of the cracked cell assay, and Dr. Suzie J. Scales for helpful comments on the manuscript.
This work was supported in part by Conte Center Grant MH48108. The costs of publication of this article were defrayed in part by the payment of page charges. This article has been marked “advertisement” in accordance with 18 U.S.C. Section 1734.
7058
References
- Chen YA, Duvvuri V, Schulman H, Scheller RH (1999) Calmodulin and Protein Kinase C Increase Ca2+-stimulated. Secretion by Modulating Membrane-attached Exocytic Machinery. Journal of Biological Chemistry 274: 26469-26476.
- https://www.nobelprize.org/nobel_prizes/medicine/laureates/2013/advanced-medicineprize2013.pdf
- https://pharmaceuticalintelligence.com/2013/09/10/synaptotagmin-functions-as-a-calcium-sensor-how-calcium-ions- regulate-the-fusion-of-vesicles-with-cell-membranes-during-neurotransmission/.
- https://pharmaceuticalintelligence.com/biomed-e-books/series-a-e-books-on-cardiovascular-diseases/ perspectives-on-nitric-oxide-in-disease-mechanisms-v2/.
- https://pharmaceuticalintelligence.com/2013/12/18/physiologist-professor-lichtstein-chair-in-heart-studies-at-the-hebrew-university-elected-dean-of-the-faculty-of-medicine-at-the-hebrew-university-of-jerusalem/.
- https://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary- arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/.
- Atrioventricular (AV) Conduction Disease (block): Human Mutations affecting the Voltage Clock .
- Genetics of Conduction Disease: Atrioventricular (AV) Conduction Disease (block): Gene Mutations – Transcription, Excitability, and Energy Homeostasis .
- Genomics & Genetics of Cardiovascular Disease Diagnoses: A Literature Survey of AHA’s Circulation Cardiovascular Genetics, 3/2010 – 3/2013 .
- Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for CardiomyocyteRyanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses .
- https://pharmaceuticalintelligence.com/2013/12/21/genetic-polymorphisms-of-ion-channels-have-a-role-in-the- pathogenesis-of-coronary-microvascular-dysfunction-and-ischemic-heart-disease/.
- https://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/.
- https://pharmaceuticalintelligence.com/2013/08/26/role-of-calcium-the-actin-skeleton- and-lipid-structures-in-signaling-and-cell-motility/.
- https://pharmaceuticalintelligence.com/2013/09/02/renal-distal-tubular-ca2-exchange-mechanism-in-health-and-disease/.
- https://pharmaceuticalintelligence.com/2013/09/08/the-centrality-of-ca2-signaling-and-cytoskeleton-involving-calmodulin-kinases-and-ryanodine-receptors-in-cardiac-failure-arterial-smooth-muscle-post-ischemic-arrhythmia-similarities-and-differen/.
- https://pharmaceuticalintelligence.com/2013/12/23/calmodulin-and-protein-kinase-c-drive-the-ca2-regulation-of-hormone-and-neurotransmitter-release-that-triggers-ca2-stimulated-exocytosis/.
- https://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/.
- https://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias- and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/.
- https://pharmaceuticalintelligence.com/2013/09/12/disruption-of-calcium-homeostasis-cardiomyocytes-and-vascular- smooth-muscle-cells-the-cardiac-and-cardiovascular-calcium-signaling-mechanism/.
- https://pharmaceuticalintelligence.com/2013/09/16/calcium-channel-blocker-calcium-as-neurotransmitter-sensor-and- calcium- release-related-contractile-dysfunction-ryanopathy/.
- https://pharmaceuticalintelligence.com/2013/09/10/synaptotagmin-functions-as-a-calcium-sensor-how-calcium-ions-regulate-the- fusion-of-vesicles-with-cell-membranes-during-neurotransmission/.
- https://pharmaceuticalintelligence.com/2013/11/01/sensors-and-signaling-in-oxidative-stress/.
- https://pharmaceuticalintelligence.com/2013/12/21/genetic-polymorphisms-of-ion-channels-have-a-role-in-the-pathogenesis-of- coronary-microvascular-dysfunction-and-ischemic-heart-disease/.
- https://pharmaceuticalintelligence.com/2013/04/14/reversal-of-cardiac-mitochondrial-dysfunction/.
- https://pharmaceuticalintelligence.com/2013/11/09/calcium-signaling-cardiac-mitochondria-and-metabolic-syndrome/.
- https://pharmaceuticalintelligence.com/2013/04/14/mitochondrial-dysfunction-and-cardiac-disorders/.
- https://pharmaceuticalintelligence.com/2013/04/14/mitochondrial-metabolism-and-cardiac-function/.
- https://pharmaceuticalintelligence.com/2013/04/14/chapter-5-mitochondria-and-cardiovascular-disease/.
- https://pharmaceuticalintelligence.com/2013/02/03/mit-scientists-on-proteomics-all-the-proteins-in- the-mitochondrial-matrix-identified/.
- https://pharmaceuticalintelligence.com/2012/11/14/mitochondrial-dynamics-and-cardiovascular-diseases/.
- https://pharmaceuticalintelligence.com/2012/10/28/mitochondrial-damage-and-repair-under-oxidative-stress/.
- https://pharmaceuticalintelligence.com/2012/09/16/nitric-oxide-has-a-ubiquitous-role-in-the-regulation- of-glycolysis-with-a-concomitant-influence-on-mitochondrial-function/.
- https://pharmaceuticalintelligence.com/2012/08/01/mitochondrial-mechanisms-of-disease-in-diabetes-mellitus/.
- https://pharmaceuticalintelligence.com/2012/07/09/mitochondria-more-than-just-the-powerhouse-of-the-cell/.


