Azharul Islam*1,2,3,4, Suraj Abraham 3,5,6,7 and Anthony P Moran8
1Division of Pathological Biochemistry, Department of Biomedical Sciences, School of Life Sciences, azharul.islam@usask.ca
2Division of Medical Biochemistry, Department of Pathophysiological and Therapeutic Sciences, Faculty of Medicine, Tottori University, Yonago, Japan
3Department of Anatomy and Cell Biology, College of Medicine, University of Saskatchewan, Saskatoon, Canada, S.A: abrahams@tbh.net
4Vaccine and Infectious Disease Organization- International Vaccine Centre (VIDO-InterVac), University of Saskatchewan, Saskatoon, Canada
5Department of Chemistry and Biochemistry, Faculty of Science, University of Regina, Regina, Canada
6Department of Experimental Pathology and Laboratory Medicine, University of Saskatchewan, Saskatoon, Canada
7Thunder Bay Regional Research Institute, Thunder Bay, Canada
8Department of Microbiology, National University of Ireland, Galway, Ireland, A.P.M: anthonypmoran @eircom.net
*Corresponding Author:
Dr. Azharul Islam
Rm 133, Vaccine and Infectious Disease Organization- International Vaccine Centre
University of Saskatchewan, 120 Veterinary Road, Saskatoon, SK S7N 5E3
Canada
Telephone: +1-306-966-7494
Fax: +1-306-966-7478
E-mail: azharul.islam09@gmail.com,azharul.islam@usask.ca
Introduction
Guillain-Barre syndrome (GBS) is an autoimmune disorder of peripheral nerves and is characterized by ascending paralysis. In spite of several studies due to its clinically importance and worldwide distribution, the etiology of this disease is still not clear. This syndrome was first described in 1892 by Sir William Osler as “acute post infectious polyneuritis” [1]. There are three different extreme patterns of clinically defined GBS, as described by van der Meche [2]. It was observed that two third of the GBS patients developed the syndrome following a pathogenic infection. The pathogens that been ascribed to leading to this syndrome include bacteria such as Cam pylobacter jejuni, Mycoplasma pneumoniae; viruses such as hepatitis B virus, cytomegalovirus, various varicella-zoster virus, Epstein-Barr virus, rubeola and human immunodeficiency virus [3-7]. Interestingly, certain studies have suggested that vaccines against rabies, influenza, and swine flu vaccine may also cause GBS-associated conditions [8-10]. However, in response to the study by Hurwitz et al (1981), Lehmann et al (2010) suggested that there was no clear association between GBS and immunization [11]. Their justification was based on rationale that the Hurwitz et al (1981) study lacked a longitudinal epidemiological data involving both active or passive surveillance, monitoring the vaccine safety similar to the one following mass immunization programmes during and after the 2009 H1N1 pandemic [11].
The recent systematic review by Poropatich et al (2010) further strengthened the findings of previous reports on Campylobacter jejuni (C. jejuni) infection as the most recognized causative factor of GBS [12]. All the available reports have suggested C. jejuni to be a potential predictor of poor outcome in patients suffering from GBS, as this pathogen induce a severe autoimmune response leading to greater axonal damage [12-18]. This is in lines with the findings shared by scientists of different disciplines and clinicians from all over the world in the workshop on “Development of GBS following Campylobacter infection” in Bethesda, Maryland on August 26- 27, 1996. Until now, no effective measures have come into effect to regulate C. jejuni-mediated GBS. However, certain learned school of thought believes that an effective vaccine development programme may provide a unique and successful preventive measure against C. jejuni-mediated GBS. They have been partially successful in this direction; however a safe and efficient vaccine programme for humans is still elusive. In this review, we briefly describe the C. jejuni-induced GBS, critically review the molecular mimicry hypothesis of C. jejuni mediated GBS, explore potential vaccine development approaches and discuss the impeding factors involved in the pursuit of safe and efficient vaccine development.
Guillain Barre Syndrome (GBS): symptoms and clinical spectrum
As mentioned earlier in this report that GBS is a neurological disease characterized by ascending paralysis, which can lead to respiratory muscle compromise and even death. Several reports have suggested a multitude of symptoms associated with GBS. These symptoms include muscle weakness and dysesthesias in the legs, which may subsequently spread to the arms and upper body. The spectrum of GBS can be described at least three levels: the clinical level, the level of antecedent infections and the level of immunologic disturbances [2,19,20]. The spectrum of clinical symptoms ranges from classical, largely motor, acute inflammatory demyelinating polyneuropathy (AIDP) to purely motor form called acute motor axonal neuropathy (AMAN), acute sensory neuropathy (ASN), and acute motor and sensory axonal neuropathy (AMSAN) and to the Miller Fisher syndrome (MFS) manifesting opthalmoplegia, ataxia and areflexia. [20]
Epidemiology and Economic impact of C. jejuni associated GBS
C. jejuni infection is much more common in developing countries than in developed countries. In almost all reported cases, males were commonly affected by GBS than females [19,67]. The age distribution is bimodal, with peaks in elderly as well as young adults between 15 and 30 years old and elderly [21]. The incidence is lower in children than in adults, with the highest incidence in the elderly [22]. The youngest patient reported was a 2 years old girl and the oldest patient was 83 years old woman [9]. Summer time outbreaks of GBS with primary axonal involvement (acute motor axonal neuropathy) are well described in China and also reported in Mexico, Spain and Korea. Familial cases of GBS are almost unknown [29]. The US Centers for Disease Control and Prevention estimates that there are 1000 cases of C. jejuni infection per 100,000 every year [23]. The number of symptomatic C. jejuni infections has been estimated to be ~2.5 million per year in the United States. The incidence of reported infections is slightly higher in males than in females [24]. The annual percentage of C. jejuni-associated GBS cases among the total GBS population in Japan was found to be in the range of 35-59% in 1990-1996, with the male to female ratio for all GBS patients was 2.2:1 [25].
Currently, the world statistics on economic burden of Campylobacter- associated GBS is not available. However, the epidemiological reports on Campylobacter-associated GBS suggest a significant economic burden to the healthcare system within the countries of European Union, United States of America and Australia [26]. In United States alone, the total medical costs associated with Campylobacter-associated GBS, is estimated to be $57.7-$420.5 million/year. According to the survey conducted in USA during 1995, the total lifetime medical and lost productivity costs due to Campylobacter-associated GBS have further inflated the estimates to $247.3-$1,799.2 million each year [16].
C. jejuni and the route of transmission in humans
C. jejuni is a microaerophilic, gram-negative, non-sporeforming bacterium with a characteristic S-shaped or spiral morphology. C. jejuni transmission to humans occurs by oral route and direct contact with infected animals, especially pets [23]. The C. jejuni infection is zoonotic and the main route of infection is through ingestion of uncooked poultry, contaminated milk and water [23].
The association of C. jejuni with GBS and the role of lipopolysaccharide mediated autoimmune responses
The typing studies proved to be critical in our understanding of the epidemiology and pathogenesis of Campylobacter-associated GBS. Currently, two major serotyping schemes are employed worldwide, viz. Lior serotyping [27] and Penner serotyping [28]. These molecular typing studies have helped us answer the questions about genetic diversity of strains associated with GBS and determine whether certain pathogenic “clones” are responsible for eliciting GBS. By molecular typing, it was possible to determine the differences between strains, even with the same serotype, and help our understanding of the association of different pathologic forms of GBS. In this section we will explore some of the reports that provided us an insight into the C. jejuni-associated GBS and the role of LPS/LOS in eliciting autoimmune responses.
The N-acetylneuraminic (Neu5Ac, sialic acid),a characteristic component of many mammalian glycolipids and glycoproteins, was discovered in C. jejuni strains by Moran et al. in 1991 [31]. A recent study have also reported the strong association of C. jejuni lipopolysaccharides in conferring virulence and colonization potential, in humans and pets, by their ability to mimic a wide range of mammalian glycans [30]. The chemical studies on C. jejuni have revealed that structures in the terminal regions of the core oligosaccharides (OSs) of specific serotypes may mimic the structures of human gangliosides. Recently, Houliston et al (2011) have also reported that the lipopolysaccharides, Paragloboside (LNnT) and P-blood group related antigens were found in up to 10 – 15% of C. jejuni strains based on the abundance of class ‘D’ and ‘F’ loci, while sialylated non-ganglioside lipopolysaccharides expression were observed in less than 5% of strains [30]. The mimicry of gangliosides by C. jejuni LPS has been proposed as a mechanism of disease whereby the immunemediated response to C. jejuni epitopes elicits the production of cross-reactive anti-ganglioside antibodies which target and damage the neural cells and tissues bearing gangliosides. The supportive findings for the hypothesis of molecular mimicry suggest that the most frequently isolated LPS/LOSs of C. jejuni associated GBS were GM1 and GD1a-bearing structures [32] and that the anti-GM1 antibodies are the most frequently observed antibodies in GBS. In addition, other C. jejuni LPS/ LOSs bear structural resemblances to that of GD3, GM2, GM3, and GT1a gangliosides. In addition, there is a strong, combined clinical and serologic evidence that suggests antiganglioside antibodies are significantly associated with GBS [33]. So it has been hypothesized that GBS may arise as a result of the production of antibodies to C. jejuni LPS due to molecular mimicry, cross-reaction with gangliosides or other structure present in peripheral nerves [32]. The serological studies appear to consistently support and contest the molecular mimicry hypothesis. However, studies have documented high prevalence of antibodies to C. jejuni in the serum of patients with GBS [17] as well as high prevalence of antibody (IgG anti-GM1) to C. jejuni in the serum of GBS patients in acute phase of the illness [34]. Interestingly, these antibodies were not detected in patients with C. jejuni enteritis that was not followed by GBS [35-37]. In addition, patients with the pure motor form of GBS had more anti-GM1 antibodies present in their blood stream, preceding C. jejuni infection, as opposed to patients with other neurological diseases or normal control [38]. These contrary findings may be an understatement of the molecular mimicry, as triggering factors for GBS initiation, suggesting that a highly complex mechanism of antibody generation in GBS patients. Oomes et al (1995) demonstrated that anti-ganglioside antibodies in GBS sera recognized surface epitopes on whole C. jejuni bacteria in a strain-specific fashion [39]. While Schwerer et al.(1995), and Moran et al (1996) showed that IgA antibodies from the C. jejuni-infected patients recognized GM1 and C. jejuni HS:2, HS:4, and HS:19 LPS [40-41].?Serum IgG from a patient who developed GBS after parenteral administration of gangliosides cross-reacted with the serotype HS: 2, HS: 4, HS: 19 and HS: 41 LPSs and this was absent in a preceding infection with C. jejuni. This clearly demonstrates that gangliosides and LPS from C. jejuni GBS-associated serotypes may cross-reactive.
Earlier studies including Hughes and Rees (1997) reported with a strong statement that while C. jejuni infection is not the only etiologic factor, however it is responsible for about one-fourth of all reported cases of GBS [9]. Moreover, Kaldor and Speed (1984) found that 38% of patients with GBS have had a recent C. jejuni infection [13]. In support of this study, Kuroki et al. (1991) and Yuki et al. (1992) have shown that a specific Penner’s serotype (PEN) PEN19 (HS: 19 or O: 19) was frequently isolated from GBS patients [14,15]. The strongest association of C. jejuni O:19 infection and GBS is also reported by Lang et al. (1997) [16]. Besides the clonality of HS: 19, a clonal population of HS:19 strains, reported by Nachamkin et al. (2001) and the great diversity in thermostable antigens of C. jejuni may suggest that the species were unique in the microbial world [66]. Initial studies on the cross-reactivity of anti-ganglioside antibodies with C. jejuni LPSs demonstrated that anti-GM1 antibodies reacted with C. jejuni HS: 19. The presence of GT1a and GD3-like epitopes in HS: 19 LPS were also confirmed serologically. Moreover, IgM anti-GM1 monoclonal antibodies (mAbs) from patients with chronic motor neuropathy were found to react with LPS of C. jejuni HS: 4, HS: 19 and HS: 50 serostrains. In addition, these exclusive antibody reaction with the core OS were demonstrated, providing us the first conclusive evidence that it is the core OS of LPS, not the lipid A or polysaccharide moiety, to which these antibodies bind [16,21]. High levels of anti-GQ1b antibodies were detected in most patients suffering from Miller Fisher syndrome (MFS), a variant of Gullian-Barre Syndrome [43]. There are reports suggesting the cross-reactivity of GQ1b ganglioside and LPS/LOSs from C. jejuni HS: 2 and HS: 23 strains [64]. However, no neuropathy associated LPS were chemically characterized to date that bears resemblances to a full GQ1b structure. The finding of GD2/GD3 ganglioside mimicry in a C. jejuni HS: 23 may be relevant because GD3 bears the terminal trisaccharide Neu5Aca2- 8NeuAca2-3Gal, and this is identical to the terminal of GQ1b ganglioside, a moiety that is repeated internally. Thus, the repeated finding of ganglioside epitopes in C. jejuni LPS/LOS by their cross-reactivity with GBS anti-ganglioside serum may be interpreted as further justification of the findings that molecular mimicry may determine the neuritogenicity of C. jejuni.
Molecular mimicry of gangliosides by C. jejuni LPS/LOS moieties
Penner and Aspinall (1997) classified the LPS antigens into 3 major classes, Class 1- complete LPS molecule; sialylated core oligosaccharide with covalently linked O chain, (O:4, O:19, O:23 and O:36), Class 2-lipooligosaccharide( LOS) type molecule, sialylated core oligosaccharide(OS); no O chain (O:1,O:2) and Class 3-nonsialylated core OS with polysaccharide polymer not linked to core oligosaccharide, (O:3). In addition, further structural analysis studies have shown that the outer core of C. jejuni O:19 LPS, including those from GBS-associated isolates, contain terminal tetra- and pentasaccharide moieties identical to those of GM1 and GD1a gangliosides, respectively (see Figure 1) [32,44-47]. In addition, the terminal hexasaccharides and trisaccharides of the LPS from some GBS-associated C. jejuni O:19 isolates were observed to have molecular mimicry to GM1, GT1a and GD3 gangliosides [46, 64].
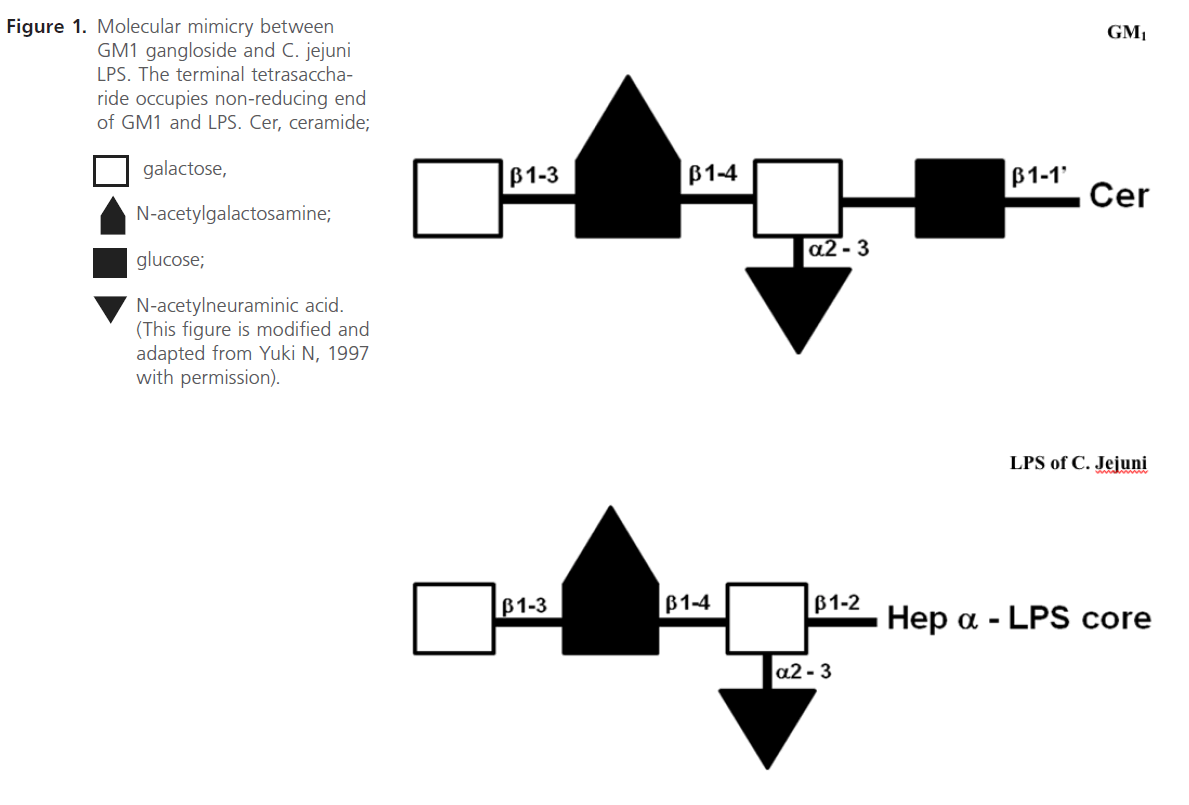
Figure 1: Molecular mimicry between GM1 gangloside and C. jejuni LPS. The terminal tetrasaccharide occupies non-reducing end of GM1 and LPS. Cer, ceramide
Mechanism of the development of GBS after C. jejuni infection
In this section we will elaborate on the mechanism involved in the development of C. jejuni associated GBS. This is based on the earlier studies on the molecular mimicry between the surface components of peripheral nerves (gangliosides) and infectious agents isolated from patients of GBS. Yuki et al (1992) showed that the purified LPS contained galactose and N-acetylgalactosamine(GalNAc) and N-acetylneuraminic acid (NeuAc), which are sugar components of GM1 ganglioside. Also the terminal structure (Gal b1-3 GalNAc b1-4 [NeuAc a2-3] Gal b) is identical to the terminal tetrasaccharide of the GM1 ganglioside (Figure. 1) [15]. Yuki (1997) placed the hypothesized that this molecular mimicry between gangliosides and lipopolysaccharides (LPSs) of C. jejuni plays the vital role in the pathogenesis of GBS [32]. GBS may arise as a result of the production of antibodies to C. jejuni LPS, which through molecular mimicry results in generating antibodies against GM1, these then cross-reacts with gangliosides or other structures present in the peripheral nerves (see Figure 2). In the same report, his research team has also shown the mechanism of antibody production against ganglioside epitope and the role of T helper cells in the production of anti- GM1 antibody [32]. Molecular mimicry between LPS of C. jejuni and GM1 ganglioside and the presence of a hyaluronic acid-like repeat unit of C. jejuni LPS helps this GM1 -like LPS to induce the production of the IgG anti- GM1 antibody and the subsequent development of GBS (Figure 1) [32,48,45]. Specific Penner serotype PEN19 (O:19 or HS:19) was most frequently isolated from GBS patients [14,15]. Isolation of LPS from O: 19 showed that the LPS has the GM1 epitope [32,49]. It has been reported that presence of anti-ganglioside antibody in blood after C. jejuni infection in GBS patients and bovine ganglioside infection may also cause GBS [49].
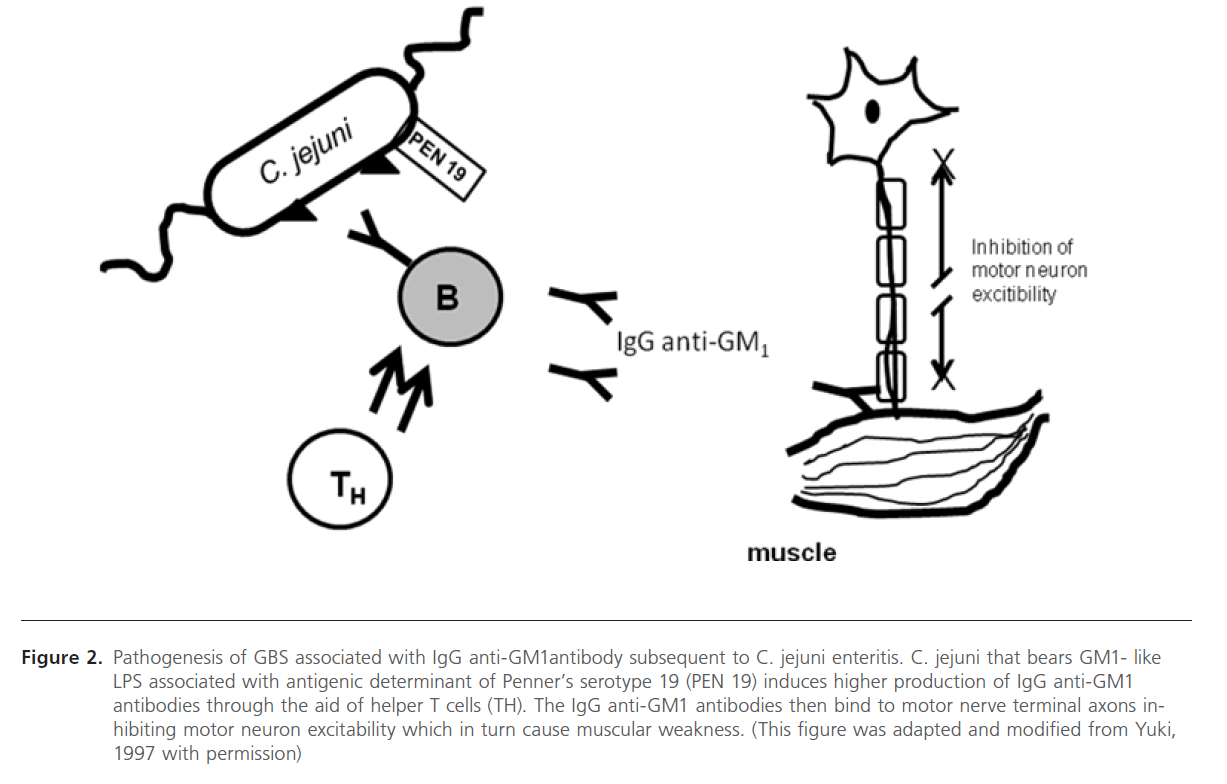
Figure 2: Pathogenesis of GBS associated with IgG anti-GM1antibody subsequent to C. jejuni enteritis. C. jejuni that bears GM1- like LPS associated with antigenic determinant of Penner’s serotype 19 (PEN 19) induces higher production of IgG anti-GM1 antibodies through the aid of helper T cells (TH). The IgG anti-GM1 antibodies then bind to motor nerve terminal axons inhibiting motor neuron excitability which in turn cause muscular weakness. (This figure was adapted and modified from Yuki, 1997 with permission)
In contravening the hypothesis of Yuki 1997, Moran and Prendergast (1998) argued specifically that high expression of the hyaluronic-like O chain may be more important for the development of GBS rather than the molecular mimicry per se [51]. Moran and Prendergast (1998) examined the expression of the O chain in 2 enteritis and 2 GBS isolates of C. jejuni O:19. They showed that the chemical liberation and purification of O chains gave higher yields from LPS of GBS than of enteritis-associated isolates [51]. In addition, Moran and O’Malley (1995) showed that the LPS core of a C. jejuni O:19 isolate from a patient with enteritis without GBS, exhibited mimicry of GM1 and GD1a oligosaccharides. Moreover, the molecular mimicry is not restricted to C. jejuni O:19, isolates of other C. jejuni serotypes obtained from GBS patients exhibit mimicry of GM1 and other gangliosides [ 41]. Thus they proclaimed that since both the GBS-and enteritis-associated isolates in their study exhibited ganglioside mimicry, differences in the extent of expression of the hyaluronic acid-like O chain, may play the vital role in the development of C. jejuni-associated GBS.
However, a seminal report by Yuki et al (2004) supporting his previous hypothesis on molecular mimicry and GBS initiation in an animal model was published [50]. In their study, when the Japanese white rabbits were sensitized with C jejuni lipooligoasaccharides containing GM1 epitope, it induced the production of pathogenic autoantibodies that can lead to peripheral neuropathy similar to GBS-associated condition. The same study also found indirect evidence to the fact that the nature of GM1-oligosaccharide structure may be important in the development of GBS. It is this finding that may have some relevance to the earlier report of Moran et al (1996) in which the difference in the extent of expression of oligosaccharide structure in C jejuni associated GBS rather than molecular mimicry mechanism postulated by Yuki et al (1991) that form the basis of GBS initiation. In lines with Moran et al (1996), Caporale et al (2006) appeared to suggest that only 1/1000 of C jejuni enteritis developed GBS, also contesting the hypothesis that molecular mimicry between glycolipid antigens expressed in C jejuni and human peripheral nerve may induce GBS [52]. However, Caporale et al (2006) emphasized on the importance of host-related factors in the development of the disease and proposed an alternate hypothesis that the polymorphism of CD1 molecule, a MHC-like glycoproteins specialized in capturing and presenting the glycolipid to antigen-specific T-Cells, may determine the susceptibility to develop GBS. However, another research group could not conclusively prove this hypothesis as they found no correlation between CD1 polymorphism, antecedent infection and GBS [53]. In spite of the several studies that support and disproves the molecular mimicry hypothesis, the common consensus is that an intricate highly complex molecular pathway in lines with molecular mimicry may be involved in C jejuni initiated GBS. However, the recent advances in glycobiology, proteomic, kinomic and genomic analysis method, along with a more multidisciplinary approach may help determine the exact mechanism of C. jejuni-associated GBS and other GBS-related conditions.
Current treatment or prevention measures
So far, the possible symptomatic treatments of C. jejuni-associated enteritis include extensive antibiotic based therapy, mechanical ventilation, plasma exchange (PE) and intravenous immunoglobulin injection (IVIG). The antibiotics are only effective during the primary stages of the infection for the symptomatic treatment of diarrhoeal illness. However, they are not effective in preventing GBS following an antecedent infection. PE and IVIG treatment were reported to be equally effective in hastening the recovery from GBS. Though PE is found to carry greater risk of side-effects and the American Academy of Neurology (AAN) does not recommend combining both these treatment options. However, studies have shown that there are differences between the treatment effects of PE and IVIG within a cluster of patients who suffer from pure motor syndrome. In these group of patients, IVIG treatment with anti-GM1 antibody or C. jejuni antibody were more effective than PE treatment alone [37,38].
Other possible preventive measures from infection with C. jejuni and GBS induction include avoiding uncooked poultry, contaminated water, unpasteurized milk, direct contact with pets, infected person, maintaining proper sanitation and taking precaution while traveling to Campylobacter endemic areas. However, successful vaccine development may still be an unique and effective preventive measure against C. jejuni associated GBS.
C. jejuni vaccine development, strategies and major constraints
The development of vaccine against C. jejuni may be feasible due to number of epidemiological reports, its economic impact and current lack of effective treatment. However, the development of vaccine may be complicated by the tremendous antigenic diversity of C. jejuni and by the fact that the protective epitopes are yet to be clearly defined.
It is widely believed that for a vaccine to be effective against an enteric agent such as C. jejuni, it must be able to stimulate intestinal immunization [54-57, 68]. Historically, several approaches to develop an oral based vaccine were explored.
Live attenuated vaccines: Using genetic approach to develop a live attenuated Campylobacter vaccine is an attractive approach [58]. But this approach is complicated by the paucity of information on pathogenesis and physiology of this organism. Interestingly, Buckley et al (2010) demonstrated that this approach could be possible, as they tested this approach by vaccinating Light Sussex chickens with Salmonella enterica serovar Thyphimurium DeltaaroA vaccine expressing the C jejuni amino acid binding proteins CjaA, Peb1A, GlnH or ChuA as a plasmid-borne fusion to the C-terminus of fragment C of tetanus toxin (TetC-CjaA, TetC-Peb1A, TetC-GlnH or TetCChuA) [59]. They found that vaccine with DeltaaroA strain of a TetC fusion to Peb1A (TetC-Peb1A) elicited protection against intestinal colonisation by C. jejuni.
Sub-unit vaccines: Two Campylobacter antigens, flagellin and a protein called PEB1, have been suggested as subunit vaccine candidates for use either as purified recombinant proteins or by expression in carrier vaccine strains, such as live attenuated Salmonella or Shigella species [60,61] . The antigenic diversity of Campylobacter flagellins, coupled with the fact that these eubacterial proteins are glycosylated, may make the development of flagellin subunit-based vaccines highly problematic, as several studies (as discussed earlier) have shown how glycosylated proteins such as hyaluronic acid may elicit different immune responses in humans.
The other protein that has been suggested as potential subunit-based vaccine target is PEB1, a highly immunogenic protein conserved among C. jejuni isolates that has also been suggested to function as an adhesion to eukaryotic cells [62]. As more is understood about the pathogenesis of Campylobacter species, it is likely that additional conserved subunit targeted proteins will be identified.
Killed WC vaccines: Inactivated microorganisms offer several advantages as potential vaccines for mucosal immunization [61]. They should enhance interactions between their surface and mucosal lymphoid tissues. As vaccines, they are inexpensive to produce and possess multiple antigens that can be important for conferring protection. The formulations can be modified to enhance protection against prevalent serotypes over time as is done for influenza virus vaccines. The presentation of multiple antigens may be particularly important for pathogen like Campylobacter species, for which protective antigens are yet to be known or these antigens are not available in recombinant forms. The Enteric program at Navy Medical Research Institute (NMRI) have studied the hypothesis that a killed WC vaccine against Campylobacter species could be safe, immunogenic, and protect against disease, particularly if combined with an effective mucosal adjuvant , such as E.coli heat-labile toxin (LT) [18,63].
Future exploration
Current animal models of Campylobacter enteritis with ensuing GBS may help understand the pathophysiological mechanism of GBS and may help pave the way to define or discover protective epitopes that confer C. jejuni’s virulence capacity, the killed WC (whole cell) vaccine of C. jejuni 19 may be used to study vaccine development approaches. The molecular mimicry hypothesis/theory has been proven by Yuki and his team using animal models. However it is absolutely necessary that a through structural studies of surface antigens of all causative agents for GBS in human neuronal cell gangliosides is still to be determined. A large-scale investigation is imminent to establish the hypothesis that the high expression of hyaluronic like O chain may play an important role in the development of C. jejuni associated GBS, however the genetic diversity of both host immune system and antigenic factors of the pathogens is yet to be established.
Interestingly, the development of GBS is rare in children < 2 years of age despite their susceptibility to C.jejuni infection. This may indicate that particular maturation of the immune system or neural antigens or receptors are needed for GBS to manifest itself. Since, the trisaccharide is a feature that distinguishes neuropathic from non-neuropathic strain and it is conceivable that the trisaccharide could be an important epitope in pathogenesis [42]. Hence, more investigation is needed to determine the acquisition of the enzyme α-2,-8- sialyltransferase and its activation and regulation. Since O:19 (PEN19 or HS-19) is the main target of GBS neuropathic manifestation because of its ability to generate anti- GM1 antibodies, clonality potential and its reported involvement with GBS being maximal [33]. However, the presence of anti- GM1 antibodies is not only concentrated with C. jejuni alone [33] but also in presence of other infectious agents such as CMV, M .pneumoniae as reported by Hao et al. [65]. Therefore, the comparative structural studies of O:19 antigen with others may correlate the both hypothesis discussed earlier. Further research is necessary to clarify the mechanism of immunemediated nerve damage which is still poorly understood.
Concluding remarks
A concrete multidisciplinary approach incorporating advance proteomic, kinomic and genomic techniques may help further explore the molecular mimicry hypothesis and characterize the C. jejuni LPS/LOS, the genetic diversity of the organism, and host immune components that trigger autoimmune response following infection. Though Campylobacter vaccine development will be a unique measure to prevent C. jejuni-induced GBS. A common and clearly defined antigenic determinant that facilitates development of a stable vaccine against C. jejuni-induced GBS is yet to be characterized. This may hinder the development of safe and effective vaccines against C. jejuni infection in humans.
Acknowledgements
The authors are grateful to Dr. Debabrata Biswas, Department of Animal and Avian Sciences, University of Maryland for his fruitful discussion and critical reading of this manuscript.
Conflict of interests
The authors declare that they have no conflict of interest
6447
References
- Osler W (1892) The principles and practice of medicine. Appleton and Co, New York: Page 777-778, 538, 541
- Van der Meche FGA, Visser LH, Jacobs BC, Endtz HP, Meulstee J and Van Doorn PA (1997) Guillain-Barre syndrome: multifactorial mechanisms versus defined subgroups. J Infect Dis 176: S99
- Dowling PC (1981) Role of infection in Guillain-Barre syndrome: laboratory confirmation of herpesvirues in 41 cases. Ann Neurol 9 (suppl): 44-55
- Glaze DG (1992) Guillian-Barre syndrome. In: Feigen RD, Cherry JD, eds. Text book of pediatric infectious diseases. Vol 1. 3rd ed. Philadelphia: WB Saunders: Pg.464-474
- Sanders EA, Peters AC, Gratana JW, Hughes RA (1987) Guillain-Barre syndrome after varicella-zoster infection. Report of two cases. J Neurol 234: 437-439
- Tsukada N, Koh CS, Inoue A, Yanagisawa N (1987) Demyelinating neuropathy associated with hepatitis B virus infection. J Neurol Sci 77: 203-216
- Thornton CA, Latif AS, Emmanuel JC (1991) Guillain-Barre syndrome associated with human immunodeficiency virus infection in Zimbabwe. Neurol 41: 812-815
- Huruwitz ES, Schonberger LB, Nelson, DB, Holman RC (1981) Guillain- Barre syndrome and the 1978-1979 influenza vaccine N Engl J Med 304: 1557-1561.
- Hughes RAC, Rees JH (1997) Clinical and epidemiological features of Guillain-Barre syndrome. J Infect Dis 176: S92
- Stowe J, Andrews N, Wise L, Miller E (2009) Investigation of the temporal association of Guillain-Barre syndrome with influenza vaccine and influenza like illness using the United Kingdom General Practice research database. Am J Epidemiol 169: 382-388
- Lehmann HC, Hartung HP, Kieseier BC, Hughes RA (2010) Guillain- Barre syndrome after exposure to influenza virus. Lancet Infect Dis 10:643-51
- Poropatich KO, Fischer Walker CL, Black RE (2010) Quantifying the Association between Campylobacter Infection and Guillain-Barré Syndrome: A Systematic Review J Health Popul Nutr; 28: 545–552
- Kaldor J, Speed BR (1984) Guillain-Barre syndrome and Campylobacter jejuni. Br Med J 288: 1867-1870
- Kuroki S, Haruta T, Yoshioka M, Kobayashi Y, Nukina M, Nakamishi H. (1991) Guillain-Barre syndrome associated with Campylobacter infection. Pediatr Infect Dis 10: 149-151
- Yuki N, Sato S, Fujimoto S, (1992) Serotype of Campylobacter jejuni, HLA, and the Guillain-Barre syndrome. Muscle Nerve 15: 968-969
- Lang DR, Allos BM, Blaser MJ (1997) Workshop summary and recommendations regarding the development of Guillain-Barre syndrome following Campylobacter infection. J Infect Dis 176: S198
- Allos BM (1997) Association between Campylobacter infection and Guillain-Barre syndrome. J Infect Dis 176: S125
- Hadden RD, Gregson NA. (2001) Guillain-Barré syndrome and Campylobacter jejuni infection. Symp Ser Soc Appl Microbiol. 30(Suppl):S145–54.
- Hughes RAC (1990) Guillain-Barre syndrome. Heidelberg, Germany: Springer-Verlag
- van der Meche FGA, Van Doorn PA (1997) The current place of highdose immunoglobulins in the treatment of neuromuscular disorders. Muscle-Nerve 20: 136-147
- Nachamkin I, Allos BM, Ho T (1998) Campylobacter species and Gullian-Barre Syndrome. Clin Microbiol Rev, 11: 555-567
- Kaplan JE, Katona P, Hurwitz Es, Schonberger LB (1982) Guillain-Barre syndrome in the United States, 1979-1980 and 1980-1981. Lack of an association with influenza vaccination. JAMA 248: 698-700
- Kendell EJ, Tanner EI (1982) Campylobacter enteritis in general practice. J Hyg 88: 155-163
- Allos BM, Blaser MJ (1995) Campylobacter jejuni and the expanding spectrum of related infections. J Clin Infect Dis 20: 1092-1101
- Saida T, Kuroki S, Hao Q, Nishimura M, Nukina M, Obayashi H (1997) Campylobacter jejuni isolates from Japanese patients with Guillain- Barre syndrome. J Infect Dis 176: S129
- Buzby JC, Roberts T, Allos BM (1997) Estimated Annual costs of Campylobacter-Associated Guillain-Barré Syndrome. Agricultural Economic Report No. 756. https://www.ers.usda.gov/publications/ aer756/AER756.PDf, Retrieved on 27 May 2011
- Lior H, Woodward DL, Edgar JA, Laroche LJ, Gill P (1982) Serotyping of Campylobacter jejuni by slide agglutination based on heat-labile antigenic factors. J Clin Microbiol 15: 761-768
- Penner JL, Hennessy JN (1980) Passive hemagglutination technique for serotyping Campylobacter fetus subsp. jejuni on the basis of soluble heat-stable antigens. J. Clin Microbiol 12: 732-737
- McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, Wu HS, Zhaori G, Liu Y, Jou LP, Liu TC, Gao CY, Mao JY, Blaser MJ, Mishu B, Asbury AK (1993) Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol 33: 333-342
- Houliston RS, Vinogradov E, Dzieciatkowska M, Li J, St Michael F, Karwaski MF, Brochu D, Jarrell HC, Parker CT, Yuki N, Mandrell RE, Gilbert M (2011) The lipooligosaccharide of Campylobacter jejuni: Similarity with multiple types of mammalian glycans beyond gangliosides. J Biol Chem. 286: 12361-12370
- Moran AP, Rietschel ET, Kosunen TU, Zahringer U (1991) Chemical Characterization of Campylobacter jejuni lipopolysaccharides containing N-acetylneuraminic acid and 2,3- diamino-2,3-dideoxy-Dglucose. J Bacteriol 173:618-26
- Yuki N (1997) Molecular mimicry between gangliosides and lipopolysaccharides of Campylobacter jejuni isolated from patients with Guillain-Barre syndrome and Miller Fisher syndrome. J Infect Dis 176: S150
- Willison HJ, O’Hanlon G, Paterson G, O’Leary CP, Veitch J, Wilson G, Roberts M, Tang T, Vincent A (1997) Mechanisms of action of anti- GM1 and anti-GQ1b ganglioside antibodies in Guillain-Barre syndrome. J Infect Dis 176: S144
- Yuki N, Yoshino H, Sato S, Miyatake T (1990) Acute axonal polyneuropathy associated eith anti-GM1 antibodies following Campylobacter enteritis. Neurology 40: 1900-1902
- Kornberg AJ, Pestronk A, Bieser K, Ho TW, McKhann GM, Wu HS, Jiang Z (1994) The clinical correlates of high-titer IgG anti- GM1 antibodies.Ann Neurol 35: 234-237
- Rees JH, Gregson NA, Hughes RAC (1995) Anti-ganglioside GM1 antibodies in Guillain-Barre syndrome and their relationship to Campylobacter jejuni infection. Ann Neurol 38: 809-816
- Visser LH, van der Meche FGA, Van Doorn PA, Meulstee J, Jacobs BC, Oomes PG, Kleyweg RP (1995) Guillain-Barre syndrome without sensory loss (acute motor neuropathy): a subgroup with specific clinical, electrodiagnostic and laboratory features. Brain 118: 841-847
- Jacob BC, Van Doorn PA, Schmitz PIM, et al. (1996) Campylobacter jejuni infections and anti-GM1 antibodies in Guillain-Barre syndrome. Ann Neurol 40: 181-187
- Oomes PG, Jacobs BC, Hazenberg MPH, Banffer JRJ, van der Meche FGA (1995). Anti-GM1 IgG antibodies and Campylobacter bacteria in Guillain-Barre Syndrome: Evidence of molecular mimicry. Annal Neurol 38: 170-175.
- Schwerer B, Neisser A, Polt RJ, Bernheimer H, Moran AP (1995). Antibody cross-reactivities between ganglioside and lipopolysaccharides of Campylobacter jejuni serotypes associated with Guillain-Barre syndrome. Innate Immunity 2: 395-403
- Moran AP, Applemelk BJ, Aspinall GO (1996) Molecular mimicry of host structures by lipopolysaccharides of Campylobacter and Helicobacter spp.: implications in pathogenesis. J Endotoxin Res 3: 521-531
- Penner JL, Aspinall GO (1997) Diversity of lilopolysacchadire structures in Campylobacter jejuni. J Infect Dis 176: S135
- Paparounas K (2004) Anti-GQ1b ganglioside antibody in peripheral nervous system disorders, Pathophysiologic role and clinical relevance. Arch Neurol, 61: 1013-1016
- Pei Z, Blaser MJ (1993) PEB1, the major cell-binding factor of Campylobacter jejuni, is a homolog of the binding component in gram-negative nutrient transport systems. J. Biol Chem 268: 18,717- 725
- Yuki N, Taki T, Inagaki F, Kasama T, Takahashi M, Saito K, Handa S, Miyatake T (1993) A bacterium lipopolysaccharide that elicits Guillain- Barre syndrome has a GM1 ganglioside-like structure. J. Exp Med 178: 1771-1775
- Aspinall GO, McDonald AG, Pang H (1994) Lipopolysaccharides of Campylobacter jejuni serotype O:19 structures of O antigen chains from the serostrain and two bacterial isolates from patients with the Guillain-Barre syndrome. Biochemistry 33: 250-255
- Aspinall GO, McDonald AG, Pang H, Kurjanczyk LA, Penner JL (1994) Lipopolysaccharides of Campylobacter jejuni O:19 structures of core oligisaccharide regions from the serostrain and two bacterial isolates from patients with the Guillain-Barre syndrome. Biochemistry 33: 241- 249
- Moran AP, O’Malley DT (1995) Potential role of lipopolysaccharides of Campylobacter jejuni in the development of Guillain-Barre syndrome. J Endotoxin Res 2: 233-235
- Hansen C, Otto E, Kuhlemann K, Forster G, Kahaly GJ (1996) Glycosaminoglycans in autoimmunity. Clin Exp Rheumatol 14 (suppl 15): S59-67
- Yuki N, Sato S, Miyatake T, Sugiyama K, Katagiri T, Sasaki H (1991) Motoneuron-disease-like disorder after ganglioside therapy. Lancet 337: 1109-1110
- Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, Taguchi K, Miyatake T, Furukawa K, Kobata T, Yamada M (2004) Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligoasaccharide causes Guillain-Barre syndrome. Proc Natl Acad Sci USA, 101: 11404-11409
- Moran AP, Prendergast MM (1998) Molecular mimicry in Campylobacter jejuni lipopolysaccharides and the development of Guillain-Barre syndrome. J Infect Dis 178: 1549-51
- Caporale CM, Papola F, Fioroni MA, Aureli A, Giovannini A, Notturno F, Adorno D, Caporale V, Uncini A (2006). Susceptibility to Gullian- Barre syndrome is associated to polymorphisms of CD1 genes. J Neuroimmunol 177:112-118
- Kuijf ML, Geleijns K, Ennaji N, van Rijs W, van Doorn PA, Jacobs BC (2008). Susceptibility to Gullian-Barre syndrome is not associated with CD1A and CD1E gene polymorphisms. J Neuroimmunol 205(1-2):110- 112.
- Childers NK, Bruce MG, McGhee JR (1989) Molecular mechanisms of immunoglobulin A defense. Annu Rev Microbiol 43: 503-506
- Holmgren J, Czerkinsky C, Lycke N, Svennerholm AM (1992) Mucosal immunity: implications for vaccine development. Immunobiology 184: 157-179
- McGhee JR, Mestecky J, Dertzbaugh MT, Eldridge JH, Hirasawa M, Kiyono H (1992) The mucosal immune system: from fundamental concepts to vaccine development. Vaccine 10:75-88
- Mestecky J (1987) The common mucosal immune system and current strategies for the induction of immune responses in external secretions. J Clin Immunol 7: 265-276
- Yao R, Burr DH, Doig P, Trust TJ, Niu H, Guerry P (1994) Isolation of motile and non-motile insertional mutants of Campylobacter jejuni: the role of motility in adherence and invasion of eukaryotic cells. Mol Microbiol 14: 883-889
- Buckley AM, Wang J, Hudson DL, Grant AJ, Jones MA, Maskell DJ, Stevens MP (2010). Evaluation of live-attenuated Salmonella vaccines expressing Campylobacter antigens for control of C. jejuni in poultry. Vaccine 28: 1094-1105
- Lee LH, Burg E, Baqar S, Bourgeois AL, Burr DH, Ewing CP, Trust TJ, Guerry P (1999). Evaluation of a truncated recombinant flagellin subunit vaccine against Campylobacter jejuni. Infect Immun 67 (11): 5799-5805
- Scott DA (1997) Vaccines against Campylobacter jejuni. J Infect Dis 176 (Suppl 2): S183-S188
- Pei Z, Blaser MJ (1993) PEB1, the major cell-binding factor of Campylobacter jejuni is a homolog of the binding component in gram-negative nutrient transport systems. J Biol Chem 268:18717-25
- Baqar S, Applebee LA, Bourgeois AL.(1997). Immunogenicity and Protective efficacy of a prototype Campylobacter killed Whole-Cell Vaccine in Mice. Infect Immun 63: 3731-3735.
- Yu RK, Usuki S, Ariga T (2006). Ganglioside molecular mimicry and its pathological roles in Guillain-Barre Syndrome and related diseases. Infection and Immunity 74(12): 6517-6527
- Hao Q, Saida T, Kuroki S, Nishimura M, Nukina M, Obayashi H, Saida K (1998) Antibodies to gangliosides and galactocerebroside in patients with Guillain-Barre syndrome with preceding Campylobacter jejuni and other identified infection. J Neuroimmunol 81: 116-126
- Nachamkin I, Engberg J, Gutacker M, Meinersman RJ, Li CY, Arzate P, Teeple E, Fussing V, Ho TW, Asbury AK, Griffin JW, McKhann GM, Piffaretti JC. (2001) Molecular population genetic analysis of Campylobacter jejuni HS:19 associated with Guillain-Barre syndrome and gastroenteritis. J Infect Dis 184: 221-226
- Blaser MJ, Taylor DN, Feldman RA (1983) Epidemiology of Campylobacter jejuni infections. Epidemiol Rev 5: 157-176
- Walker RI, Clements JD (1993) Use of the heat labile toxin of enterotoxigenic Escherichia coli to facilitate mucosal immunization. Vaccine Res 2: 1-10

