Keywords
CO intoxication; Children; MRI; Mitocondrial disorder
Introduction
Carbon monoxide (CO) intoxication is the leading cause of poisoning in industrialized countries. In Italy it’s possible to estimate that CO poisoning is the cause of 6000 hospitalization/year and approximately 350 deaths per year [1]. Carbon monoxide has no colour, no odor, no taste and CO intoxication lacks any premonitory sign.
Carbon monoxide intoxication is one of the main causes of diagnostic errors in emergency medicine, because presenting symptoms are extremely aspecific and confounding [2], even more in infants: it’s often misdiagnosed as influenza ill, gastroenteritis, headache. We present a case of a toddler with a misdiagnosed CO intoxication.
Case Report
The patient is a 4-months-old boy with an uncomplicated birth history, born by natural vaginal delivery at 40 weeks of gestation, birth weight 3230 gr (50th centile), head circumference 34 cm (15th-50th centile), lenght 51 cm (50th centile), APGAR score 9/9 at 1' and 5'.
During summer holidays parents noticed an abrupt change in baby’s mood and interaction and the occurrence of recurrent episodes, with the baby shuttering eyes and stiffening, going into transitory apneas with face flushing. Moreover during the following days they observed the loss of previously acquired abilities and palpebral ptosis.
Ten days after symptoms’ onset, the boy was taken to the emergency department of a peripheral hospital. On admission he was described as stuporous, with physiological cardiac and respiratory examination, normal muscular tone, no nuchal rigor, no signs of endocranial hypertension. Maternal abuse of cocaine and cannabis was referred until third month of gestation. Subsequently the mother had successfully attended a disintoxication program.
Family history was silent. Parents described a normal physical and mental child’s development until four months of age, with head control acquired at three months of age, social smile and vocalizations.
The boy presented with persistent miosis, photomotor hyporeactive reflex, no eye contact and visual pursuit, incostant jerks of vertical nystagmus. He showed fieble reactivity to stimuli (no crying), chaotic and aimless limb movements, loss of sitting posture and head propping. Head circumference was at 5th centile. Toxicological analyses resulted negative (phencyclidine, acetaminophen, opioids, methadone, cocaine, cannabinoids, tricyclic antidepressive, benzodiazepines, barbiturates, amphetamine, methamphetamine), EEG was normal.
Blood analyses and serum chemistries were normal, as hemogasanalysis, apart from a COHb of 1.40% (range of normality 0.00-0.5%), but this element hadn’t been initially taken into account, as it was considered too low to demonstrate intoxication.
On admission to our Hospital the boy was afebrile, arterial blood pressure 86/43 mmHg, FC 110 bpm, cardio-respiratory and abdominal physical examination appeared normal. His mother presented with a severe post-partum depression and was scarcely reliable in taking care of her baby.
US-encephalography, brain CT scan, auditory evoked potentials, serum and urine amino acid analyses, urinary organic acids and abdominal US were normal.
Twenty-three days after clinical onset brain MRI scan showed hyper-intense area in T2 (Figure 1A and 1B) involving peri-aqueductal gray matter, quadrigeminal bodies-appearing bulgy, thalami and lenticular nuclei, especially putamen bilaterally. Either hypothalamus and mammillar bodies, seemed to present a possible alteration. These lesions presented low signal T1 images without restriction in protonic diffusion lacking enhancement after of contrast medium administration. These multiple, symmetric lesions led to suspicion of a mitocondrial disorder, as they can be part of MRI spectrum in Leigh Syndrome. Lactic acid was initially found at 4.9 mmol/L (normal range 0.2-2.2 mmol/L), and its elevation supported the diagnostic hypothesis of mitochondrial disorder, but values gradually decreased in the following days.
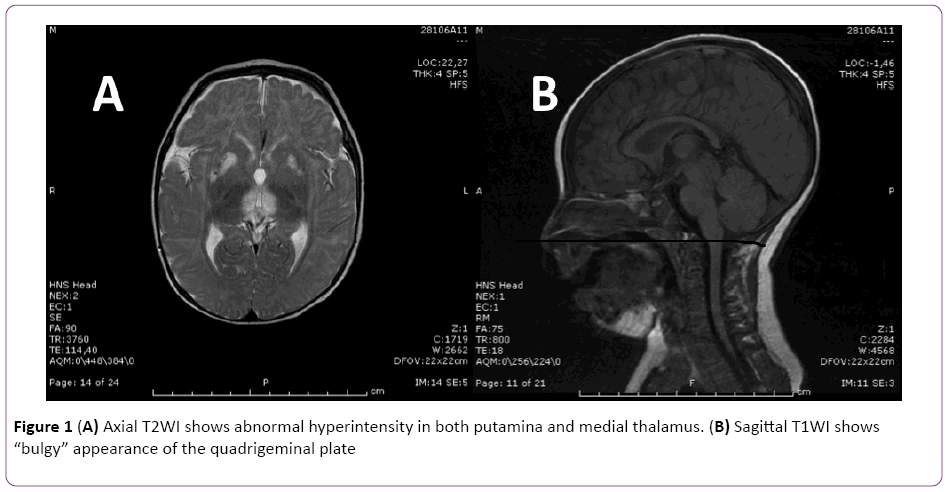
Figure 1: (A) Axial T2WI shows abnormal hyperintensity in both putamina and medial thalamus. (B) Sagittal T1WI shows “bulgy” appearance of the quadrigeminal plate
Two weeks later a H MR spectroscopy was performed confirming the presence of lactic acid consistent with a mitochondrial disorder (Figure 2A and 2B). Conversely it showed a marked reduction of previous lesions in periacqueductal grey matter, quadrigeminal bodies, medial talamic and lenticular nuclei bilaterally, especially putamen, turning the differential diagnosis towards an acute toxic disorder.
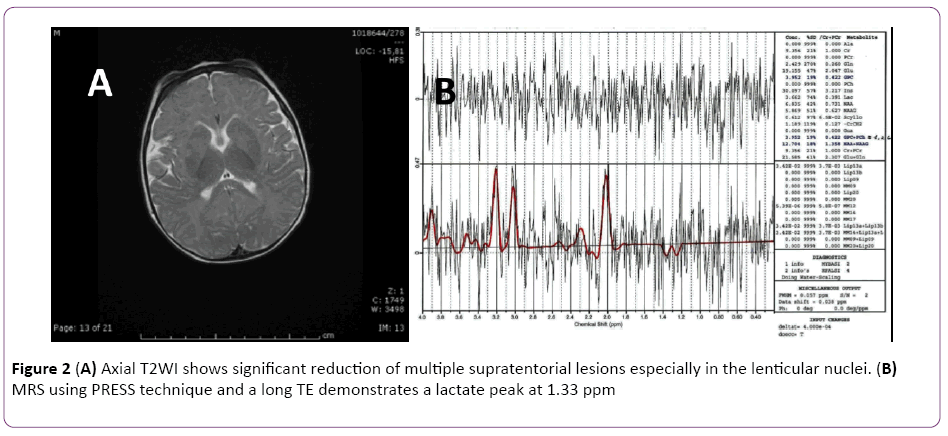
Figure 2: (A) Axial T2WI shows significant reduction of multiple supratentorial lesions especially in the lenticular nuclei. (B) MRS using PRESS technique and a long TE demonstrates a lactate peak at 1.33 ppm
Evaluation on venous hemogasanalysis at 45 days from the onset, resulted in a COHb level of 0.3%.
At the same time symptoms were gradually receding: waking hours progressively increased, along with the the boy’s visual pursuit abilities. Motor abilities, formerly lost, were gained anew.
These data supported a final diagnosis of CO intoxication, whose motivating factor is still undefined (burnt moka pot? homicide-suicide attempt? Use of water pipe?).
Six months later, at the age of 12 months, our patient underwent a brain MRI that resulted in a complete normalization of previously documented lesions (Figure 3). Head circumference was 44 cm, 5th centile. Evalutation of psichomotor development with Griffith’s Mental Development Scales, showed delayed achievement of developmental milestones comparing with peers.
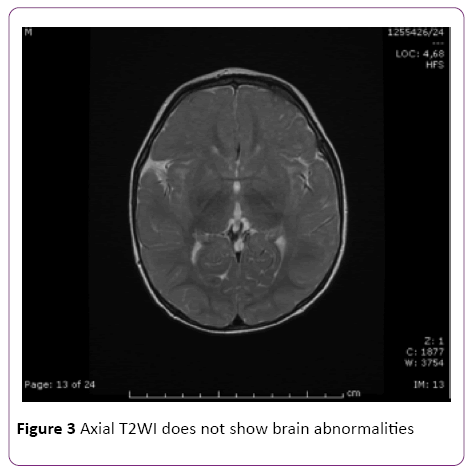
Figure 3: Axial T2WI does not show brain abnormalities
Discussion
Any process altering brain metabolism can lead to basal ganglia damage: their role in regulating extrapyramidal motor activity (receiving projections from almost every region of cerebral cortex) demands a real high energy supply, in the form of ATP; consequently they need rich blood perfusion and high concentration of trace metals. Moreover basal ganglia metabolic rate is particularly increased in children. Many chronic inborn errors of metabolism (as mitocondrial disorders), degenerative or dysmyelinating diseases could involve basal ganglia with focal damage.
Signs and symptoms displayed by our patient are extremely nonspecifics and could be ascribed to many neurological diseases of childhood. In this case it has not been possible to define with precision carbon monoxide’s source because of really scarce reliability of our patient’s parents. Despite that, many arguments depose in favor of a carbon monoxide intoxication’s hypothesis: first of all progressive and complete resolution of symptoms and signs in about four weeks beside a progressive resolution of brain MRI abnormalities, not possible in a progressive degenerative mitochondrial disease as it was suspected in first instance and second the evidence from literature that basal ganglia findings at MRI are highly specific for carbon monoxide intoxication [3].
Additionally COHb levels are above physiological limits and are to be considered pathological especially in infants: children show various symptoms at COHb levels considered asymptomatic for adults [4,5]. First determination of COHb had been taken about two weeks after symptoms’ onset and this may explain why they were slightly above the upper limit. In addition, as remarked in many papers, COHb levels are currently not considered the data directing therapeutic decisions (i.e. treatment with high flow mask oxygen or HBOT). The main issues are patient’s history and clinical presentation, in particular the degree of neurological impairment [6]. The correlation between COHb measurements and clinical symptoms and signs is still a matter of debate in literature [7]. In our patient, two weeks after hospital’s admission, COHb levels has returned broadly in physiological range (0.3%).
Hemoglobin affinity is 200 to 250 times greater for CO than for O2. CO binds competitively to hemoglobin and thereby decreases the percentage of oxyhemoglobin available in bloodstream. CO shifts hemoglobin dissociation curve to the left with a reduction of oxygen cessation to tissues. It also binds avidly to myoglobin and mitochondrial cytochromes a and a3, interfering with electron transport chain function: this results in inhibition of cellular respiration and it leads to tissue hypoxia, particularly in high demand organs such as brain and heart. Both mitocondrial disorders and acute CO intoxication inhibit oxidative phosphorylation (OXPHOS) so the ultimate consequence, basal ganglia damage, is identhical. Carbon monoxide also stimulates leukocyte adherence to brain microvasculature, and the following conversion of endothelial cell xanthine dehydrogenase to xantine oxidase by leukocyte proteases [8]. Free oxygen radicals generated by xantine oxidase lead to brain lipid peroxidation [9]. These later effects of CO damage probably explain the clinical presence of “delayed neurological sequelae” (DNS) [10] that is a delayed onset of neuropsychiatric symptoms after a lucid period, distinctive feature of CO poisoning.
CO injury seems to be more pronounced during early developmental stage: a metabolic stress that would be considered mild or moderate in an adult could have a worse impact in a child, probably because of higher minute ventilation, higher oxygen metabolism, and immaturity of nervous system in infancy. Fetal hemoglobin, persisting in a significant percentage for several months postpartum, has a higher affinity for CO than adult hemoglobin does. This means that the same level of inhaled percentage of CO that could determine no damage in adults could be very dangerous in children, and even more in an infant or fetus [6].
In our case exact timing of symptoms’ onset and informations about an eventual lucid free period after onset are missing: therefore it’s not possible to recognize the presence of DNS, according to the history collected through patient’s parents. Children have a much lower incidence of DNS than adults (2.8-10%) [10] and DNS are uncommon in patients treated with normobaric oxygen [11]. Anyway there are few studies about DNS especially in children, also because of a selection bias of the studied population: there’s a huge number of children that do not come into hospital because of blurred symptoms [12,13]. DNS usually develops 2 weeks to 1.5 months after the acute phase of CO intoxication in 12 to 50% of adults [14,15].
Conclusions
Many elements in this case had been confounding: clinical history was unclear because of important maternal postpartum depression, CO origin wasn’t determined, COHb was quite low, even if out of normal range, and the boy presented with a low head circumference. Diagnosis was finally established thanks to: neuroradiological findings, indicative for CO poisoning with normalization after six months; COHb levels turning normal after ten days; gradual regression of neurological symptoms, especially ophthalmological ones. In front of an acute onset of neurological symptoms, Co intoxication should always be taken into account.
8701
References
- Locatelli C, Casagranda I, Coen D, Demattè P (2001) Gestione e trattamento del paziente con intossicazione acuta da monossido di carbonio: linee guida. The Online Journal of Anesthesiology Italia 6.
- Barret L, Danel V, Faure J (1985) Carbon monoxide poisoning, a diagnosis frequently overlooked. J Toxicol Clin Toxicol 23: 309-313.
- Hopkins RO, Fearing MA, Weaver LK, Foley JF (2006) Basal ganglia lesions following carbon monoxide poisoning. Brain Inj 20: 273-281.
- Klasner AE, Smith SR, Thompson MW, Scalzo AJ (1998) Carbon monoxide mass exposure in a pediatric population. Acad Emerg Med 5: 992-996.
- Baker MD, Henretig FM, Ludwig S (1988) Carboxyhemoglobin levels in children with nonspecific flu-like symptoms. J Pediatr 113: 501-504.
- Martin JD, Osterhoudt KC, Thom SR (2000) Recognition and management of carbon monoxide poisoning in children. Clin Ped Emerg Med 1: 244-250.
- Hampson NB, Hauff NM (2008) Carboxyhemoglobin levels in carbon monoxide poisoning: do they correlate with the clinical picture? Am J Emerg Med 26: 665-669.
- Thom SR (1993) Functional inhibition of leukocyte B2 integrins by hyperbaric oxygen in carbon monoxide-mediated brain injury in rats. Toxicol Appl Pharmacol 123: 248–256.
- Thom SR (1990) Antagonism of carbon monoxide-mediated brain lipid per- oxidation by hyperbaric oxygen. Toxicol Appl Pharmacol 105: 340–344.
- Choi IS (1983) Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol 40: 433-435.
- Meert KL, Heidemann SM, Sarnaik AP (1998) Outcome of children with carbon monoxide poisoning treated with normobaric oxygen. J Trauma 44: 149-154.
- Cho CH, Chiu NC, Ho CS, Peng CC (2008) Carbon monoxide poisoning in children. Pediatr Neonatol 49: 121-125.
- Lonati D, Giampreti A, Locatelli CA (2012) Carbon monoxide poisoning in children. Pediatr Neonatol 53: 75.
- Thom SR, Taber RL, Mendiguren II, Clark JM (1995) Delayed neurological sequelae following carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med 25: 474-480.
- Kurt F, Bektas Ö, Kalkan G, Öncel MY, Yakut HI, et al. (2013) Does age affect presenting symptoms in children with carbon monoxide poisoning? Pediatr Emerg Care 29: 916-921.

