Manzano Palomo S1, Rioja M2, J. Kulisevsky3, Jiménez-Escrig A4*
1Servicio de Neurologia, Hospital Clínico San Carlos, Madrid, Spain.
2Servicio de Medicina Nuclear, Hospital Ramon y Cajal, Madrid, Spain.
3Servicio de Neurologia, Hospital Santa Cruz y San Pablo, Barcelona, Spain.
4Servicio de Neurología, Hospital Ramon y Cajal, Madrid, Spain.
- *Corresponding Author:
- Dr. Jiménez-Escrig
Servicio de Neurología Hospital Ramon y Cajal 28034 Madrid.
Fax: 34 1 3369016
Email: adriano.jimenez@hrc.es.
Keywords
Parkinson´s disease, PINK1, DJ1, PARKIN, LRRK2, α-synuclein.
Introduction
The majority of PD cases are sporadic although genetic forms are increasingly recognized. In familial PD of Mendelian inheritance there are twelve forms reported so far. Focussing on those with recessive transmission, three forms have been reported to date. PARK2, secondary to Parkin mutations was the first described, occurring worldwide.[1] These patients have clinical manifestations very early, at about 20-30 years of age. PARKIN mutations are responsible of 49% of familial early onset cases and some sporadic cases with early onset.[2] The other two recessive forms, PARK6 and PARK7, were reported in two European families previously mapped to chromosome 1p36. In PARK6, the responsible gene was identified as the PTEN-induced putative kinase 1, or PINK1 kinase (1p35-36),[3] and PARK7 as DJ-1.[4] Mutations in PARKIN are usually rearrangements, deletions or insertions, although point mutations have been also reported. Mutations in DJ-1 are an exon deletion, point deletions and a substitution Leu166Pro in exon 54 while so far in PINK1 only point mutations have been reported. However, according to simulation and epidemiological studies the genetic cause of at least 50% of familial cases of PD is still unknown,5 and possibly the number of causative genes in PD may be up to twenty. The finding of these genes and the study of the function of the codified protein and its role in the degenerative pathways of PD is a major method to unravel the cause of this disease. To achieve this objective, the study of families with this disorder has been a major tool. In these cases, the study begins with the clinical description of these families and their genetic investigation to rule out the involvement of genes known to date.
We report the clinical and genetic data on a Spanish family with a late onset autosomal recessive Parkinson´s disease and the molecular genetic exclusion of the previously reported genes causative of the recessive forms of PD (Parkin, DJ1 and PINK1), of mutations in the coding region of α-synuclein gene and of the common mutation G2019S in LRRK2 gene.
Patients and Methods
The kin group as presently collected is shown in Figure 1. They were originally from a village in a mountain range that is geographically isolated. Affected cases were directly evaluated by at least one of the authors. Evaluation consisted of a physical and neurological examination, and genealogical history to ascertain additional affected family members. In identified cases, we tested serum glucose and creatinine, hepatic function, erythrocyte sedimentation rate, and serum caeruloplasmin and copper levels without finding abnormalities. The diagnosis of PD was based on the presence of two of two or more of the three cardinal clinical symptoms (tremor, ridigity or akinesia) during the period of the study, as well as the absence of signs or symptoms of atypical parkinsonism or secondary causes, plus the presence of clinical response to levodopa therapy. Age at onset was defined as the age when the individual first complained of neurological symptoms, for example rest tremor. Retrospective data on neurological disease in deceased family members was obtained from their living relatives. Affected subjects were videotaped according to a standard protocol and blood samples were drawn under informed consent for DNA extraction. Total genomic DNA was isolated from blood by Quiagen Maxiprep DNA extraction kit. For homozygosity exclusion, we selected two markers flanking each gene (Parkin: D6S1035F, D6S980F; DJ-1: D1S548F, D1S1612; PINK1: D1S1571F, D1S478F) using the Taiwan Polymorphic Database. These markers where amplified by polymerase chain reaction (PCR) and run in an ABI342 to determine their size. Homozygosity for a mutation in a candidate gene was excluded if both flanking markers of the gene where heterozygous. LRRK2 G2019S mutation was ruled out by PCR amplification and sequencing of exon 41 of the LRRK2 gene. Mutations in the coding region of alpha synuclein were ruled out as well by sequencing of α-synuclein exons[1-6].
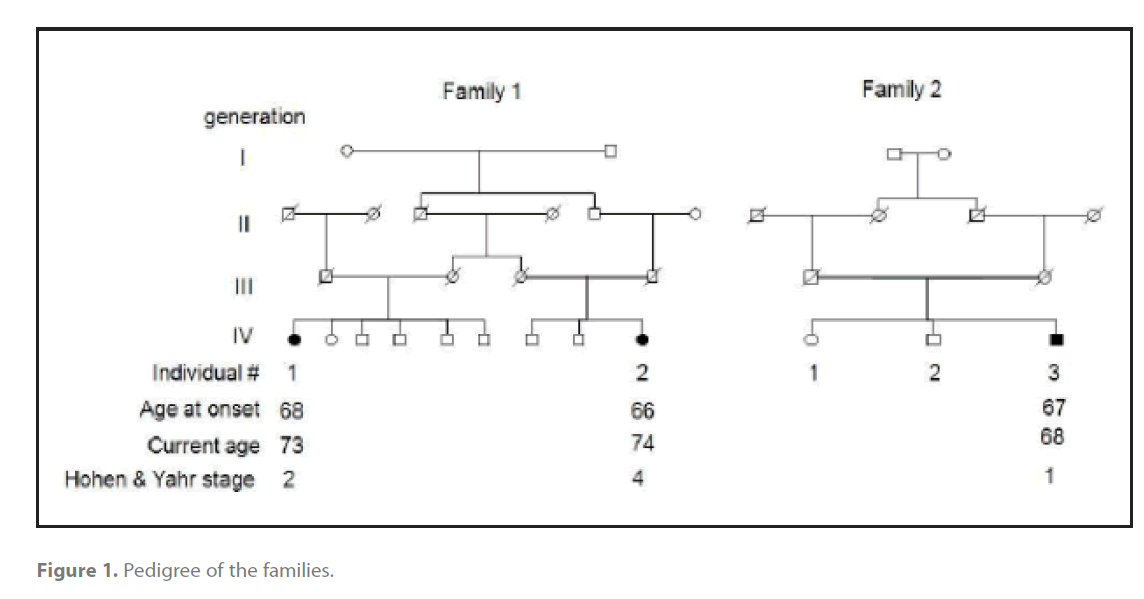
Figure 1. Pedigree of the families.
In order to confirm the presynaptic involvement of the disease striatal uptake of iodine-123-betaIoflupane SPECT (Datscan®, GE) was assessed in two cases. Patients received a single intravenous injection of 123I-Ioflupane (111–185 MBq DaTSCAN, Amersham Health, Buckinghamshire, UK) and underwent a SPECT scan four hours later. sequences. In addition, the postsynaptic D2-receptor density was determined by 123I-iodobenzamide (IBZM) SPECT.
Results
The pedigree diagram in figure 1 depicts ages, age of onset and Hohen and Yahr stage of disease. Their inheritance pattern is autosomal recessive with a common founder because the three affected members are only from the last generation and it is a highly inbred pedigree considering that two of them are the child of first cousins, that they were born in the same region, a genetic isolate in a mountain range and that they share the family name. These three patients have a slowly progressive Parkinson disease with good levodopa response. Age at onset ranged 66-68 years, and duration of disease ranged 1-7 years. Dementia, pyramidal, cerebellar or autonomic disturbances were not present. Since there are not deceased cases we lack data about neuropathology.
The iodine-123-betaIoflupane SPECT (Datscan®, GE) of cases Family 1 III-1 (Figure 2A) and family 2 III-3 (Figure 2B) showed a reduced uptake in putamen and caudate nucleus bilateral although asymmetrical, demonstrative that a presynaptic lesion was responsible for the clinical picture. The striatal IBZM uptake was normal in Family 2 case III-3 (Figure 2D), whilst case Family 1 III-1 of had a reduced basal ganglia/frontal cortex ratio (Figure 2C).
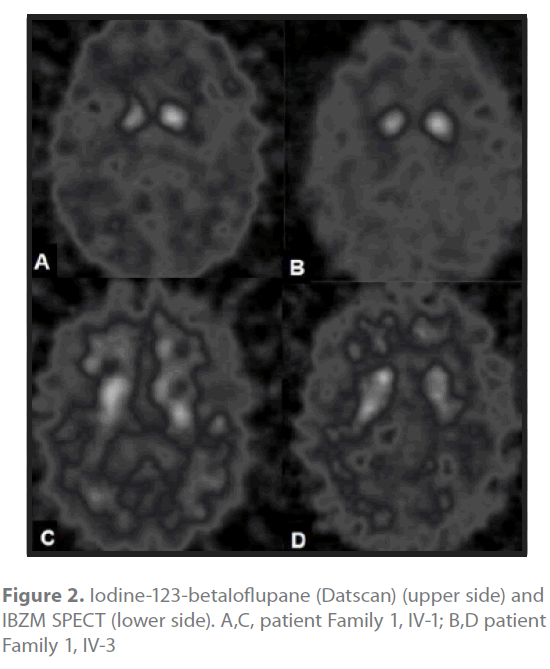
Figure 2. Iodine-123-betaIoflupane (Datscan) (upper side) and IBZM SPECT (lower side). A,C, patient Family 1, IV-1; B,D patient Family 1, IV-3
Discussion
These patients represent a new form of autosomal recessive familial Parkinson disease that contrariwise to the previous reported forms is characterized by a late clinical onset. This is an interesting finding in the genetics of PD considering that all recessive forms reported so far have early onset, [6-9] although there are a few families described with an atypical late onset PD as the cases reported herein.[7]
The molecular study ruled out the recessive genes described so far. In addition, the common mutation in LRRK2 G2019S was also ruled out considering that this mutation is very frequent is South Europe populations and that mutations in this gene have an incomplete penetrance that can resemble a recessive inheritance. We also ruled out mutations in the coding region of α-synuclein, despite mutations so far reported in this gene have an autosomal dominant transmission, because of the key role of this gene in PD.
The presynaptic origin of the lesion was confirmed by iodine-123- betaIoflupane SPECT DaTSCAN demostrating a degeneration of the nigrostriatal pathway,[10] likewise to idiopatic late onset PD, with the anteroposterior gradient typically seen in idiopathic PD[11] that conveys the preferential involvement of the posterior striatum because of the preferential cell loss in the ventrolateral tier of the pars compacta of substantia nigra which projects to posterior putamen rather than to caudate.[12] The presence of a reduced density of postsynaptic receptors in one case (family 1, case III-1) is not irreconcilable with idiopathic PD since previous studies using 11C-raclopride PET in patients with PARK2 showed reduced binding showed reduced binding in the striatum[13]. Besides, this patient was on dopamine agonists therapy that was discontinued only 48 hours prior to scanning, a washout phase too short to reverse all the possible down-regulation of postsynaptic D2 receptors known to occur during long-term dopaminergic therapy.[14]
Late onset disease genetically determined is not simple to detect for reasons such as mortality by unrelated diseases or accident before reaching the age of penetrance, comorbidity and geographical separation of affected relatives. In addition, if the disease is recessive the probability of two or more siblings affected is lower, increasing difficulties to detect these familial forms. The presence of late onset together with a recessive pattern of transmission may justify the complex genetics attributed to neurodegerative diseases, PD among others.[15] However, naming it complex genetics points only to our lack of understanding of the genetics of a disease, and this term is abandoned and replaced by another the very moment the mechanism of the disease is unravelled.
2234
References
- Ishikawa A, Tsuji S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology. 1996; 47:160-166
- Lucking CB, Abbas N, Durr A et al. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson’s Disease and the French Parkinson’s Disease Genetics Study Group. Lancet. 1998; 352:1355-1356
- Valente EM, Bentivoglio AR, Dixon PH et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001; 68:895-900
- Bonifati V, Oostra BA, Heutink P. Linking DJ-1 to neurodegeneration offers novel insights for understanding the pathogenesis of Parkinson’s disease. J Mol Med. 2004; 82:163-174
- Lüking CB, Dürr A, Bonifati V, and et al. Association between earlyonset Parkinson´s disease and mutations in the parkin gene. N.Engl.J Med. 342, 1560-1567. 2000.
- Klein C, Hedrich K, Wellenbrock C et al. Frequency of parkin mutations in late-onset Parkinson’s disease. Ann Neurol. 2003; 54:415-416
- Valente EM, bou-Sleiman PM, Caputo V et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304:1158-1160
- Bonifati V, Rohe CF, Breedveld GJ et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005; 65:87-95
- Kitada T, Asakawa S, Hattori N, and et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605-608. 1998.
- Catafau AM, Tolosa E. Impact of dopamine transporter SPECT using 123I-Ioflupane on diagnosis and management of patients with clinically uncertain Parkinsonian syndromes. Mov Disord. 2004; 19:1175-1182
- Khan NL, Valente EM, Bentivoglio AR et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol. 2002; 52:849-853
- Bernheimer H, Birkmayer W, Hornykiewicz O et al. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973; 20:415-455
- Hilker R, Klein C, Ghaemi M et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001; 49:367-376
- Antonini A, Schwarz J, Oertel WH et al. Long-term changes of striatal dopamine D2 receptors in patients with Parkinson’s disease: a study with positron emission tomography and [11C]raclopride. Mov Disord. 1997; 12:33-38
- Mayeux R. Mapping the new complex genetic disorders. J Clin Invest 115, 1404-1407. 2005.

