Keywords
Ca2+/cAMP; Signaling interaction/clinical research
Introduction
Nowadays, an emerging concept in clinics is the “translational research”. Basically, “translating” current knowledge from basic science to approaches for treating human diseases-from bench to bedside-is the main concept. Our discovery entitled “calcium paradox” fits properly in this concept. From basic science, we know that in mammals, increases of the concentration of free Ca2+ ions in the cytosol ([Ca2+]c) serve as a messenger signal to couple the stimulus to muscle contraction, or to neurotransmitter release, among other physiological responses [1,2]. A huge number of experiments performed since the discovery of the role of Ca2+ in the control of the heart beat [3] have set the dogma that in excitable cells, the increased Ca2+ influx by voltage-activated Ca2+ channels (VACC) elicited by depolarizing stimuli, triggers muscle contraction and the release of neurotransmitters, and hormones. Conversely, the mitigation of Ca2+ influx produced by VACC blockers causes a reduction of those responses [4,5].
The above concepts imply that the enhanced Ca2+ entry during cell depolarization and/or enhanced Ca2+ release from the sarco-endoplasmic reticulum (ER) augments the (Ca2+)c and the triggering of the contractile, or secretory responses. However, about four decades ago, a study showed that verapamil at low concentrations produced a paradoxical increase of the contractions mediated by sympathetic nerves from vas deferens [6]. On the other hand, nifedipine was recently found to paradoxically augment the exocytosis of catecholamine triggered by double-pulse depolarizations from voltage-clamped bovine adrenal chromaffin cells, another interesting model to study sympathetic neurotransmission [7]. How these two L-type VACC blockers can enhance, instead of reducing, the Ca2+-dependent responses of contraction, and secretion? We properly gave a response to this “calcium paradox” in 2013 through the Ca2+/cAMP signaling interaction [8].
Role of Ca2+/cAMP signaling interaction in clinical research
In the vas deferens, both release and postsynaptic actions of noradrenaline (NA), and other neurotransmitters such as adenosine 5´ triphosphate (ATP), depend on Ca2+ influx by VACC, and the ensuing elevations of (Ca2+) [9]. Hence, some authors found that verapamil abolished both noradrenergic and purinergic components of the sympathetically-mediated contractions of the vas deferens [10,11]. In an old report, however, it was showed that verapamil inhibited the sympathetically-mediated contractions of the rat smooth muscles (vas deferens), as predictable; nevertheless, this report also described that the low concentrations of verapamil produced a surprising increase of those contractions [6]. This paradoxical effect was corroborated in 1981 by French and Scott [12], also in the sympathetically-mediated contractions of the vas deferens. Furthermore, six years later a third study reported that verapamil and diltiazem increased the sympathetically-mediated contractions of the vas deferens; this result was attributed to an agonist effect of CCB on L-type VACC, thus increasing Ca2+ influx and neurotransmitter release [13]. Another published report (two years later) revealed that both, L- type VACC blockers and activator BAY K 8644, elicited similar increases of the sympathetically-mediated contractions of the smooth muscles (vas deferens); the authors did not provide a reasonable explanation for such paradoxical observation [14].
In a study from our laboratory, we could replicate those previous observations in the sympathetically-mediated contractions of the rat vas deferens: low verapamil concentrations produced a small increase in muscle contraction, while high CCB concentrations caused full inhibition of contractions [8]. Similar to the effects observed with high concentrations of CCB, various cAMP enhancers (e.g. the phosphodiesterase inhibitor rolipram) and adenylyl cyclase (AC) activators (e.g. forskolin) also reduced sympathetically mediated contractions; however, low verapamil concentrations in the presence of cAMP enhancers caused a significant increase of muscle contractions. The increased muscle contraction can in turn be reduced by adding an AC inhibitor, SQ22536, to lower the cytosolic cAMP level. These findings suggest that interactions in the Ca2+/cAMP signaling pathways could play a key role in explaining the "calcium paradox" as observed in the presence of CCB and cAMP enhancers [8]. Thus, these findings can dramatically impact on the cardiovascular, neurodegenerative disorders, cancer and other diseases [15-17].
Based on these findings, we have anticipated that the pharmacological modulation of the Ca2+/cAMP signaling interaction by combined use of the L-type CCB and cAMPenhancer compounds could be a novel therapeutic goal for increasing neurotransmission in neurological, and psychiatric disorders, resulted from neurotransmitter release deficit, and neuronal death [15,16]. This neuroprotector strategy opens a novel pathway for the drug development more efficient and safer for the therapy of several neurodegenerative diseases, including Alzheimer´s, Parkinson´s, amyotrophic lateral sclerosis and Huntington´sdiseases [15,16]. Figure 1 illustrates how the pharmacological modulation of the Ca2+/cAMP signaling interaction could be used in human therapy.
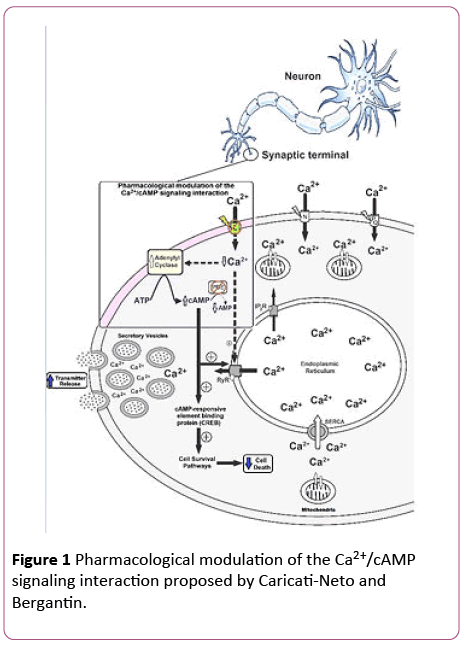
Figure 1: Pharmacological modulation of the Ca2+/cAMP signaling interaction proposed by Caricati-Neto and Bergantin.
The Ca2+/cAMP signaling interaction can be pharmacologically modulated by combined use of drugs that reduce (Ca2+)c such as CCB, and cAMP-enhancer compounds such as PDE inhibitors and AC activators. This pharmacological modulation could be a new strategy to attenuate neuronal death caused by cytosolic Ca2+ overload and to increase neurotransmitter release. L, N, PQ: Ca2+ channel types; PDE: phosphodiesterase; RyR: ryanodine receptors; IP3R: IP3 receptors; SERCA: sarcoendoplasmic reticulum Ca2+-ATPase; (+): stimulation; dotted arrow: weak effect; solid arrow: strong effect.
In fact, it was demonstrated that the prescription of L-type CCBs reduces motor symptoms, and reduces progressive neuronal death in animal model of Parkinson´s disease, indicating that L-type CCBs are potentially viable neuroprotective pharmaceuticals [18]. Intriguingly, a 1-decade study involving thousands senile hypertensive patients demonstrated that prescription of L-type CCBs reduced blood pressure, and risk of dementia, in hypertensive patients, indicating that these pharmaceuticals could be clinically used to treat neurodegenerative diseases [19]. These results for the neuroprotective effects of CCBs have been reinvestigated in thousands elderly hypertensive patients with memory dysfunction [20]. These studies concluded that patients who have taken CCBs had their risk of cognitive dysfunction decreased, such as Alzheimer´s disease [20]. These findings reinforce the idea that reduction of cytosolic Ca2+ overload produced by L-type CCBs due to blockade of Ca2+ influx could be an alternative pharmacological goal to reduce, or prevent, neuronal death in neurodegenerative diseases.
Due to involvement of the Ca2+ and cAMP signaling pathways in the regulation of the cellular differentiation process, it is possible that the pharmacological modulation of the Ca2+/cAMP signaling interaction could stimulate the cellular differentiation in stem cells [15,16]. In this case, this pharmacological strategy could be used in the cell therapy for the treatment of cardiovascular and neurodegenerative diseases.
It has been shown that the dysregulation of intracellular signaling pathways mediated by Ca2+ and cAMP participates in the cancer initiation, tumor formation, tumor progression, metastasis, invasion and angiogenesis. Thereby, proteins involved in these pathways, such as Ca2+ channels and cAMPdependent protein kinase (PKA), represent potential drugs targets for cancer therapy [17]. With this concept in mind, some studies showed that drugs able to interfere with the intracellular Ca2+ signaling, such as L-type CCB, inhibit proliferative response in different cancer cells. In addition, cAMP-enhancer compounds, such as PDE 4 inhibitors, have been proposed as potential adjuvant, chemotherapeutic or chemopreventive agents in some cancer types, including hepatocellular carcinoma [17]. Then, the pharmacological modulation of the Ca2+/cAMP signaling interaction in the cancer cells may represent a new therapeutic strategy for cancer progression.
Conclusion
The Ca2+/cAMP signaling interaction may dramatically impact on medical research and therapeutics, stimulating the development of new pharmacological strategies for the therapy of human diseases, including: cancer, cardiovascular diseases, neurodegenerative diseases, and yet cellular therapy using stem cells.
Acknowledgment
Caricati-Neto and Bergantin thank the continued financial support from CAPES, CNPq and FAPESP (Bergantin´s Postdoctoral Fellowship FAPESP #2014/10274-3).
19103
References
- Berridge MJ (2013) Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 7: 2-13.
- Berridge MJ (2014) Calcium regulation of neural rhythms, memory and Alzheimer's disease. J Physiol 592: 281-293.
- Ringer S (1883) A third contribution regarding the Influence of the Inorganic Constituents of the Blood on the Ventricular Contraction. J Physiol 4: 222-225.
- Fleckenstein A (1983) History of calcium antagonists. Circ Res 52: I3-16.
- Garcia AG, Garcia-De-Diego AM, Gandia L, Borges R, Garcia-Sancho J (2006) Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev 86: 1093-1131.
- Kreye VA, Luth JB (1975) Proceedings: Verapamil-induced phasic contractions of the isolated rat vas deferens. NaunynSchmiedebergs Arch Pharmacol 287: R43.
- Rosa JM, Conde M, Nanclares C, Orozco A, Colmena I, et al. (2011) Paradoxical facilitation of exocytosis by inhibition of L-type calcium channels of bovine chromaffin cells. Biochem Biophys Res Commun 410: 307-311.
- Bergantin LB, Souza CF, Ferreira RM, Smaili SS, Jurkiewicz NH, et al. (2013) Novel model for "calcium paradox" in sympathetic transmission of smooth muscles: Role of cyclic AMP pathway. Cell Calcium 54: 202-212.
- Burnstock G (2009) Autonomic neurotransmission: 60 years since sir Henry Dale. Annu Rev Pharmacol Toxicol 49: 1-30.
- Hidalgo A, Beneit J, Lorenzo P (1983) Effect of calcium antagonists on the response of the rat vas deferens to noradrenaline and field stimulation. Rev Esp Fisiol 39: 211-215.
- Hata F, Fujita A, Saeki K, Kishi I, Takeuchi T, et al. (1992) Selective inhibitory effects of calcium channel antagonists on the two components of the neurogenic response of guinea pig vas deferens. J Pharmacol Exp Ther 263: 214-220.
- French AM, Scott NC (1981) A comparison of the effects of nifedipine and verapamil on rat vas deferens. Br J Pharmacol 73: 321-323.
- Moritoki H, Iwamoto T, Kanaya J, Maeshiba Y, Ishida Y, et al. (1987) Verapamil enhances the non-adrenergic twitch response of rat vas deferens. Eur J Pharmacol 140: 75-83.
- Rae GA, Calixto JB (1989) Interactions of calcium antagonists and the calcium channel agonist Bay K 8644 on neurotransmission of the mouse isolated vas deferens. Br J Pharmacol 96: 333-340.
- Caricati-Neto A, Garcia AG, Bergantin LB (2015) Pharmacological implications of the Ca(2+)/cAMP signaling interaction: From risk for antihypertensive therapy to potential beneficial for neurological and psychiatric disorders. Pharmacol Res Perspect 3: e00181.
- Bergantin LB, Caricati-Neto A (2016) Challenges for the pharmacological treatment of neurological and psychiatric disorders: Implications of the Ca2+/cAMP intracellular signalling interaction. Eur J Pharmacol 788: 255-260.
- Errante PR, Caricati-Neto A, Bergantin LB (2017) Insights for the inhibition of cancer progression: Revisiting Ca2+ and cAMP signalling pathways. Adv Cancer Prevention 2: e103.
- Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 43: 364-371.
- Wu CL, Wen SH (2016) A 10-year follow-up study of the association between calcium channel blocker use and the risk of dementia in elderly hypertensive patients. Medicine (Baltimore) 95: e4593.
- Hanon O, Pequignot R, Seux ML, Lenoir H (2006) Relationship between antihypertensive drug therapy and cognitive function in elderly hypertensive patients with memory complaints. J Hypertens 24: 2101-2107.