Keywords
Imatinib; Kinase; Docking; Tyrosinase; Cancerous cells
Abbreviations
3D: Three-Dimensional; Å: Angstrom = 1.0 × 10-10 meters; aa: Amino Acid; ACE: Atomic Contact Energy; PTKs: Protein Tyrosine Kinases; ATP: Adenosine Triphosphate; TK: Tyrosine Kinases; GOLD: Genetic Optimization for Ligand Docking; FDA: Food and Drug Administration; NMR: Nuclear Magnetic Resonance; PDB: Protein Data Bank; RMSD: Root Mean Square Deviation; STI571: Imatinib Mesylate; MD: Molecular Dynamics; Hrot: Score Expressing Loss of Conformational Entropy of Ligand When it Binds to the Protein
Introduction
Kinases
Kinases are divergent and comprise one of the largest gene families. They play a crucial role in signal transduction and in cell cycle [1]. They perform their activity by causing the addition of phosphate to the substrate. They cause addition of phosphate to the amino acids tyrosine, threonine and serine residues of protein substrate [1] In eukaryotes protein kinases present a diverse family of proteins, that plays a significant part in metabolism, regulation, differentiation, transcription and also in cyto skeletal rearrangement as well as in apoptosis in addition to a wide diversity of signal in transduction processes occurring in the cell. There are total 518 protein kinases in human in which 478 constitute a single super family [2].
Tyrosine kinases
Phosphorylation of tyrosine which is one of the crucial covalent modification in multicellular organisms are caused by the protein called tyrosine kinases abbreviated as PTKs, that is involved in catalyzing the transference of phosphate from ATP to the tyrosine residues which are present on protein substrates. Tyrosine residues phosphorylation modifies enzymatic activity in addition to generating binding sites for conscription to downstream signaling proteins. Major two classes of PTKs exist in cells which are the trans membrane receptor PTKs in addition to the non-receptor PTKs. Since PTKs are serious constituents for cellular signaling pathways, but their catalytic activity is severely regulated. Over the past many years, high-resolution structures are being studied of PTKs. They are providing a molecular base to perceive the mechanisms through which receptor as well as non-receptor PTKs are being regulated [3].
Tyrosine kinases and cancer
Tyrosine kinases are essential modulators for the signaling cascade, which shows its key roles in various biological processes alike growth, metabolism, and apoptosis both intrinsic and extrinsic in addition to differentiation. Current advances have associated the part of tyrosine kinases in the pathology as well as in physiology of cancer. However, their activity is strictly regulated in case of normal cells; they can attain transforming functions owing to mutations, over expression in addition to autocrine paracrine stimulation which leads to malignancy. Constant activation of oncogenes in cancer cells could be blocked through discerning tyrosine kinase inhibitors then therefore considered as a favorable approach for new genome based therapeutics. The mechanism of activation of oncogenes and the diverse methods for inhibition of tyrosine kinase, alike small molecule inhibitors, heat shock proteins, immune conjugates, monoclonal antibodies, antisense as well as peptide drugs are studied in light of the key molecules. Angiogenesis is one of the main event in cancer development and its proliferation, for targeting angiogenesis tyrosine kinase inhibitors could be appropriately applied as a novel approach for cancer therapy [4] (Figure 1).
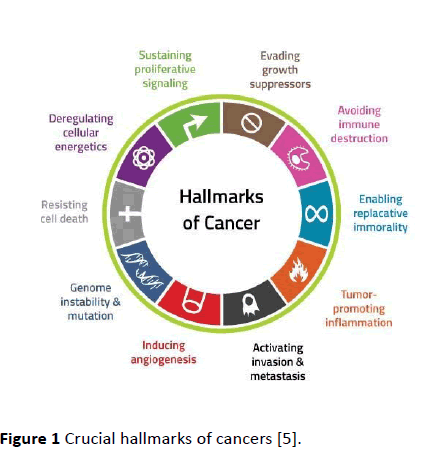
Figure 1 Crucial hallmarks of cancers [5].
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors have potential effect in the directed treatment of numerous malignancies. In clinical oncology, Imatinib was the first that was acquainting with and then it was tracked by many drugs like gefitinib, sorafenib, erlotinib, sunitinib, in addition to dasatinib. Though they have the same mode of action, i.e. competitive nhibition of ATP by binding at catalytic binding site of these tyrosine kinases, they mainly differ with each other in the range of directed kinases, their pharmacokinetics also differ in addition to adverse effects which are substance-specific. The most common adversarial effect of Imatinib is peri orbital edema. Moreover, the hematological adverse effects of many of Tyrosine Kinase Inhibitors alike anemia, thrombo penia and neutron penia, while the most commonly reported are extra-hematologic adverse side effects include edema, nausea, hypothyroidism, vomiting in addition to diarrhea. Concerning probable long term side effects, in recent times cardiac toxicity by means of congestive heart failure has been under consideration in patients who are being administered by Imatinib besides sunitinib therapy; though, this observation was most likely related to the patients selection, even though, Tyrosine Kinase Inhibitors totally seems to be actually well accepted drug class [4].
Molecular docking
Molecular docking has captured the attention of bioinformaticians for last few decades. Structure-based methods for drug designing uses the 3D structure knowledge for predicting a receptor bound with a given molecule so that the binding of ligand with a given molecule or congeneric molecules should be optimized. Docking has crucial role in structure-based methods for drug designing by putting a ligand into the pocket of active site of given macromolecule by noncovalent manner. In this way, the conformational flexibility of a molecule can be viewed by molecular docking which is otherwise very perplexing problem.
Nevertheless, of these challenges, this method has become useful tool to make new inhibitors by finding and explaining their interaction with the active site and predicting their mode of action. Molecular modeling has excelled the growing number of different X-ray and NMR structures of ligands for a given molecule and has also made the NMR and X-ray methodologies very useful in drug-designing.
In docking two major steps are involved, the prediction of exact orientation of active conformation into the active site binding pocket which is called pose and the approximation of strength of target-ligand binding interactions which is called scoring. [6].
Molecular recognition is attained by two things first is the complementarily of surface structures and second is the energetic which is usually related with slight conformational changes. The complementarities molecular surface structure can take several forms like charge-charge interaction, van der Waals' interaction, in addition to the size and shape of surfaces and the most important is hydrogen bonding [7].
Molecular docking can be done as rigid docking or by way of flexible docking. In rigid docking protein and ligand are treated as rigid structures and flexibility is not introduced in them however it is very scarcely successful. Maximum docking algorithms perform rigid docking. In flexible protein ligand docking complete flexibility in the ligand is generally induced which allow investigation of torsional degrees of freedom in process of docking. To extend this further few docking programs moreover allow partial flexibility in some residues of protein throughout docking experiment.
Protein flexibility
Flexibility as well as dynamics is protein features that are crucial for the molecular recognition process. Conformational changes into the protein which are combined with ligand binding are defined through the biophysical models which are induced fit and conformational selection. Diverse concepts are studied that include protein flexibility in protein-ligand docking in perspective of these two models. Numerous computational researches are available which confer the rationality and probable restrictions of such methods (Figure 2).
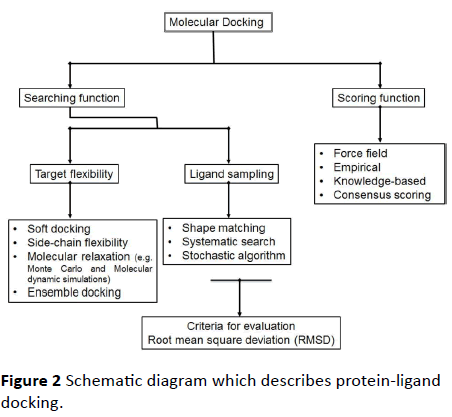
Figure 2 Schematic diagram which describes protein-ligand docking.
Soft docking
In soft docking there is a small overlap among the active site of target protein and its ligand. It is a simplest approach and is appropriate for inducing slight conformational changes. It has benefits of being effective and fast in addition to being easily implemented [7].
Side-chain flexibility
In docking the molecular interactions among the target protein and its ligand is principally directed through the side chains of amino acids of protein. By considering this method, initial efforts for integration of conformational changes in molecular docking was done by keeping the backbone of protein fixed while allowing flexibility in the side chains. Leach, which is one of the initial docking tool also operates by inducing side-chain flexibility using rotamer library [8]. In present docking algorithms, side-chain flexibility is still induced.
Molecular relaxation
In molecular relaxation method rigid docking is done in which the ligand is placed in to the active site then the side chains of receptor protein which are close to receptor are relaxed besides that the backbone of protein is also relaxed. Proteins are dynamic molecules they are not rigid thus the clatters among the ligand conformation and the active site of protein could be reduced by utilizing methods like Molecular Dynamic (MD) simulations for relaxing the complexes (1). This method is advantageous over other methods because it adds flexibility to both side chains as well as to the backbone of protein however this approach is more perplexing and needs accurate scoring functions in addition to that it is time consuming.
Ensemble docking
In ensemble docking method the flexibility in protein is induced by assembling all possible conformational changes into the structure of protein. Primarily, this approach was used for generation of an averaged energy grid. This grid was produced by joining all the energy grids which are made by known individual protein structures which are determined experimentally [9]. Docking algorithm Flex E (Claussen, Buning, Rarey and Lengauer,) utilize ensemble docking approach.
Ligand sampling
Ligand sampling is the most significant part of protein-ligand docking approach. Many improvements have been attained in that particular area of molecular docking. Usually, algorithms for ligand sampling are classified in three main types which include shape matching, systematic search and stochastic algorithms.
Shape matching
In shape matching algorithm the appropriate ligand binding pose is searched by considering molecular surface. The main aim of this algorithm is to find shape complementarily of ligand with protein binding site. This method is effective however the ligand conformation is fixed during the process [10]. Docking algorithm which use this method are DOCK [10], FRED [11] and Ligand Fit [12].
Systematic matching
This method is utilized in flexible-ligand docking. It generates all possible ligand conformations by permitting freedom of ligand rotation in all directions.
Ligand division in many fragments or in various rigid parts is done in fragmentation method. These parts are then placed into the binding site individually or all fragments are collectively placed through covalent interactions. Docking algorithms which utilize this approach are eHiTs (Zsoldos, Reid et al. 2006), DOCK [13] and FlexX [14].
Stochastic algorithms
In Stochastic algorithms at each step random changes are applied to the ligand in either rotational or translational space in addition to conformational space. There are four types of stochastic algorithms one of them is Monte Carlo (MC) algorithm.
In Monte Carlo algorithms, the approach which is used for approval or rejection of arbitrary change is determined by calculating Boltzmann probability function [15].
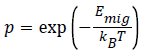
Where,
KB= Boltzmann constant.
T=Absolute temperature.
Scoring functions
In molecular docking scoring functions are utilized to evaluate the ranking of diverse plausible poses of different ligands compared to each other. There are large and constantly growing numbers of scoring functions which are accessible. They can be classified into three major categories viz. force field scoring functions; knowledge based scoring functions in addition to empirical scoring functions.
Empirical scoring functions
Hydrogen bonding, electrostatic interactions, hydrophobic interactions in addition to solvent exclusion volumes are the sort of interactions which may be involved in empirical scoring functions [16].
Preparative sets of complexes which are experimentally determined are utilized to know coefficients for the several terms. ChemScore, LigScore, LUDI,F-Score and X-Score are the examples of Empirical Scoring functions.
ΔG binding = Δ Go + ΔGh bond-Sh bond + Δ G metal S-metal + ΔG lipo S-lipo + ΔG rot H-rot
Chem-Score = ΔG binding + ΔE clash + Eint + Ecov
Shbond = Scores for hydrogen bonding.
Smetal = Acceptor-metal.
Slipo = Lipophilic interactions.
Hrot = Score expressing loss of conformational entropy of ligand when it binds to protein.
Consensus scoring
The combination of anyone of the scoring function is called as consensus scoring. There exists a conceptual difficulty in creating an association as well as in scaling of dissimilar scoring functions; in spite of the fact that it is a short coming consensus scoring approaches have shown some attainment. X-CSCORE is one of the example of consensus scoring that associates three scoring functions which include OMP, ChemScore in addition to FlexX.
Binding site
The binding site is buried in protein which is excellent for “drug ability.” The drugs of our choice which can be found or newly designed occupy this binding site. Drug is always small as compared to protein for which it is designed. The more accurately the drug is bound to the binding site, the more effective the drug is. Ligand protein binding is like “Key in a lock”. Binding site specification is the backbone of docking calculations. The success of docking depends on pharmacophore recognition. Pharmacophore site is defined as “Structural features present at receptor site on which the biological activity of a molecule is dependent.” The proper identification of pharmacophore improves the results of docking tools to a great extent.
Different algorithms for molecular docking
First designed docking tool was Dock 1.0 by Irwin Kuntz in 1982 at University of California. Now-a-days there are many docking algorithms which are available in market: AutoDock, AutoDock Vena, Flex X, Ligand Fix and comparatively new docking tools are: Glide, FRED and the latest is Surflex. Some aspects of docking tools which are commonly used for studyare concisely described below.
Glide: Friesner et al initially developed Glide [17] and then it has become a standard choice for molecular docking afterward [18]. Glide is one of the standard docking tool. It is accessible currently in Schrödinger software suite. There is a need to generate set of grids prior to docking by this tool with diverse types of fields showing geometries and characteristics of the binding site present at the given receptor. Ligand binding poses are created of ligand molecule by comprehensive sampling at the torsional space. Four major steps are involved in docking process. At first and second step, hierarchical filters are applied by program in order to search for probable locations of ligand and possible ligand poses are created through screening. At third step, ligand binding pose which was generated by screening are minimized. At last step, ligand binding poses are ranked by Emodel [19].
Gold: This program was initially developed by Jones [20]. It is currently on the rampage commercially by Cambridge Crystallographic Data Center. Gold depends on genetic algorithm (GA) to search the conformational space for the ligand. It also lets the contemplation of the conformational flexibility of many designated amino acid residues present on the protein. When three-dimensional structures are known of desired protein and ligand, their preliminary population of poses is created arbitrarily. On the basis of anticipated binding affinity all single of the population was allocated a fitness Score. ChemScore [21,22], ASP [23] and GoldScore (16,17,35) are three scoring functions which are applied in Gold for that purpose. Ranking is also done in accordance with fitness Scores [19].
Auto dock: Like Gold, AutoDock create ligand poses by using genetic algorithm. Initially Morris developed it. It uses Lamarckian version of GA, in which the conformational changes assumed by molecules subsequently in situ optimization were utilized in production of offspring poses. Like Gold active site selection was done on the basis of location of inherent ligand structure present in active site [19] (Figure 3).
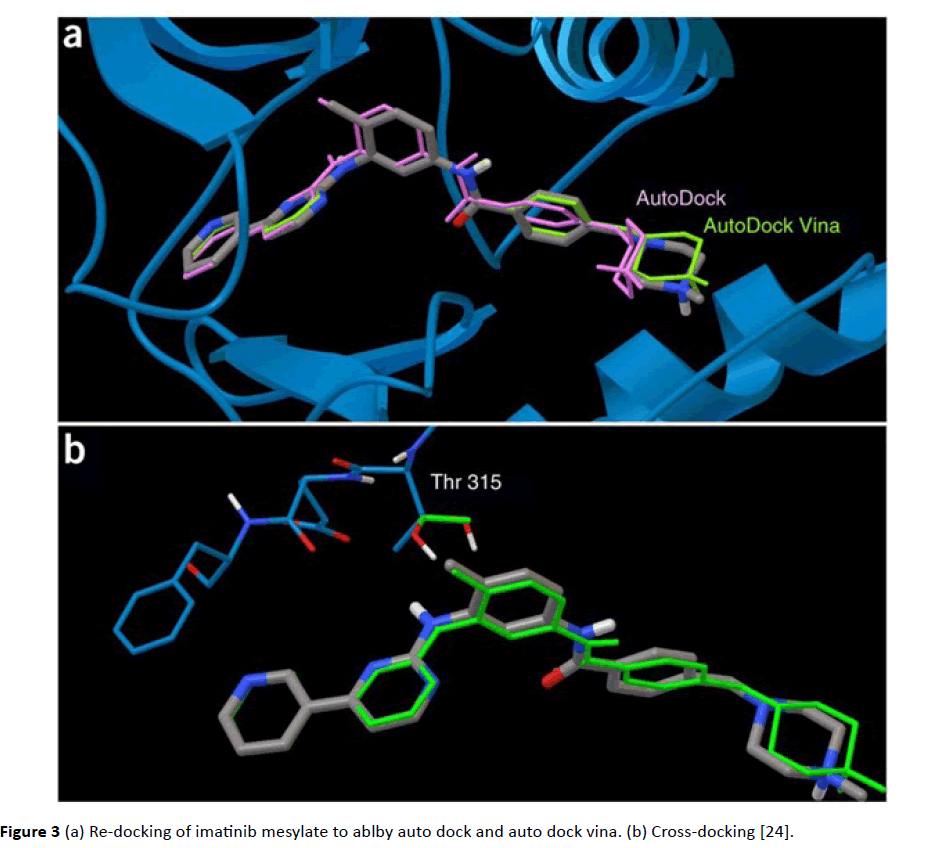
Figure 3 (a) Re-docking of imatinib mesylate to ablby auto dock and auto dock vina. (b) Cross-docking [24].
Hex server: In Hex Server molecular docking can be performed both manually as well as by online at (https:// hexserver.loria.fr/). Hex Server does not require any registration or any license agreement it is a docking server which is freely available. The working principle of Hex Server is first Fourier transform (FFT) based. In FFT based method rigid docking is done however all possible orientations are collected through searching 6D space in approximately 15 seconds. It requires the receptor as well as ligand to be in PDB format. It predicts 1000 conformations of ligands [25].
Surflex: This program was initially made by [26]. Surflex use molecular docking method which consists of two main steps. First of all “protomol” is produced [27] which is best ligand fitting to its binding site. For protomol generation on the binding site of protein three altered categories of molecular fragments which include hydrophobic group, donor group which donate hydrogen and acceptor group for hydrogen bond are placed. Moreover, their locations are enhanced for formation of best interactions with protein. Then fragments which are top-scored are collected for formation of protomol. In second step, the ideal binding pose is found by applying an incremental algorithm. Fragments of ligand are generated by breaking the ligand. Then conformations of every fragment of ligand are discovered, they are associated to analogous regions present on protomol. The fragments which are associated are estimated by comparison of steric complementarily to their binding site along with binding scores [26,28].
Patchdock: Patchdock (https://bioinfo3d.cs.tau.ac.il) is an online docking web server that operates by applying algorithm which works on the principles of shape complementarity. This algorithm is geometry-based that find transformations in order to escape steric clashes and also induce wide interface areas.
Patch dock divides Connolly dot surface presentation of ligand in various patches like flat, convex and concave. For the generation of transformations, patches are matched in accordance with complementarity and as a result candidate transformation of each is evaluated through scoring function [29] (Figure 4).
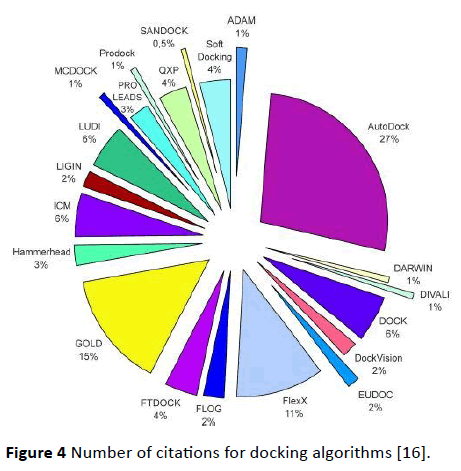
Figure 4 Number of citations for docking algorithms [16].
Molecular docking tools evaluation
Main issues in docking are (1) Docking algorithm ability of reproducing X-ray pose of required ligand which is usually small molecular weight (2) Scoring functions propensity for prediction of free energies of binding from the pose which is best-scored (3) In experiments for virtual screening the binders which are known can easily be discriminated from the molecules which are chosen randomly [30].
However, prediction and analysis of data from comparative evaluation of different docking tools is very tough job. Firstly, limited numbers of tools are available. Secondly, the comparative performances of different docking algorithms in which they are studied independently are very intermittent. Thirdly, the properties which are to be examined their quality of judgment may differ like prediction of free energy for binding, Virtual Screening Accuracy, and all possible poses quality. Fourthly, the assumption of approximation levels of most docking algorithms can be variable for example their speed may vary from few second to hours.
It has been reported few years back that docking tools have ability to predict poses of different types having variation ranging from 1.5 to 2 A0 rms [30,31]. Nevertheless, this is not proved by me while using these docking tools. Moreover, papers have been published in current years in which the performance and evaluation of different docking tools have been reported in database searching. Scoring functions accuracy have been examined in these reports, after completion of docking, thus proving the assumption that experimental poses have accurately identified by docking procedure which is success of docking procedure. This question remained still answerable that either a docking tool will influence hit rates of screening methodology in silico or not. Nonetheless, before finding answer to this question, it is more important to know the excellent docking tool accuracy for finding experimental result of ligand-protein complexes by complete and in comparative manner. Therefore, a comprehensive comparative evaluation of different docking tools is missing. Adequate data is not available about different docking tools and docking algorithms which provide us comprehensive detail about protein-ligand interactions and their binding in comparison to experimental results.
Aims and Objectives of the Study
The main objective of my study is to find independent standards for different docking tools which are widely used. The selection criteria for choosing these docking programs are:
(1) Accessibility (2) Use of file formats which are commonly used like PDB (3) they can be used easily for Virtual Screening. Four docking tools are chosen which areAutoDock, AutoDock Vena, Hex Server and Patch dock. We use different protein families and docked them with different ligands. The results here are interpreted based on ability of these docking tools in prediction of modes for binding of ligand-protein complexes in comparison to experimental poses. The comparison shows that the performance of some docking tools is constantly better from other and the relationship among binding site and excellent docking tool was evaluated. This was actually comparison of soft docking with rigid docking. Soft docking generated better results as compared to rigid docking.
Materials and Methods
Selection of protein-ligand complexes from PDB
The structures of tyrosine kinase domains especially bcr-abl were taken for building a dataset needed for present comparative analysis of docking tools. The Protein Data Bank (PDB; https://www.rcsb.org/pdb/) is distinct worldwide record for structural data set of almost all biological macromolecules [31] 5 entries were taken from Protein Data Bank (Figure 5).
Figure 5 Screenshot displaying the Home Page of Protein Data Bank form where complexes were retrieved.
The descriptive analysis of the structures taken from PDB caused the assortment of only 3 Tyrosine kinase-ligand complexes (Table 1). The segregation of only 3 entries was due to the reason that these structures were of very complex nature and they are beyond the scope of present study.
| Sr. No. |
PDB ID |
Title |
| 1 |
1iep |
Crystal Structure of the C-Abl Kinase Domain in Complex with Sti-571 |
| 2 |
1mr8 |
Migration Inhibitory Factor-Related Protein 8 from Human |
| 3 |
2GS2 |
Crystal Structure of the Active EGFR Kinase Domain |
| 4 |
1t46 |
Structural Basis for The Auto inhibition And STI-571 Inhibition of C-Kit Tyrosine Kinase |
| 5 |
1RJB |
Crystal Structure of FLT3 |
Table 1: Protein-ligand complexes selected for docking.
Describing input conformations of ligands
The prepared ligands were taken from ZINC Database. It is accessible for downloading ligands (https://zinc.docking.org) in numerous file formats which include SMILES, mol2, 3D SDF, as well as in DOCK flexibase format. Ligands were downloaded in mol2 format and then converted into PDB format by using Open Babel software because the docking tools like AutoDock Vina, Hex Server and Patchdock require that ligand should be in PDB format. Energy minimization was not accomplished in order to reserve the coordinates which are experimentally tested (Figure 6).
Figure 6 Zinc database from where ligands were taken.
Preparation of target proteins for docking
Protein files were prepared in Pymol to remove water molecules and ligands present in the complex. Hydrogen atoms were added besides Kollman and Gestiger charges were also added to make protein flexible.
Docking with auto dock
Both Protein and ligand was uploaded in PDB file format. PDBQT files (Protein. PDBQT, Ligand. PDBQT) were prepared for both protein as well as for ligand (Figures 7 - 12).
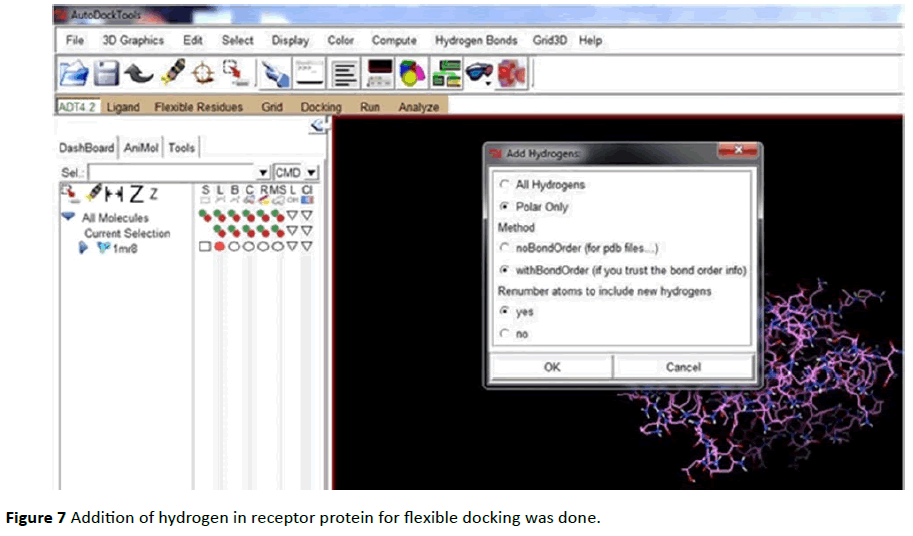
Figure 7 Addition of hydrogen in receptor protein for flexible docking was done.
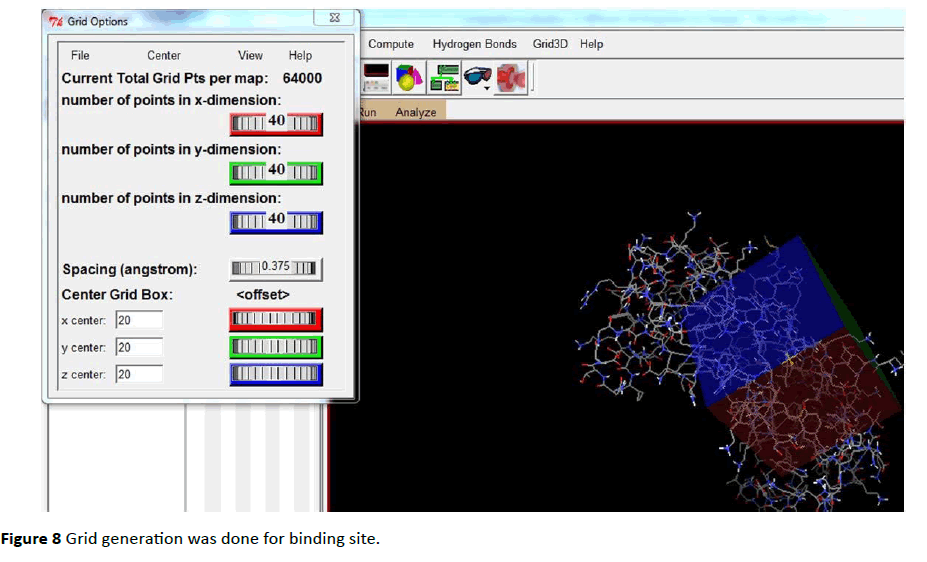
Figure 8 Grid generation was done for binding site.
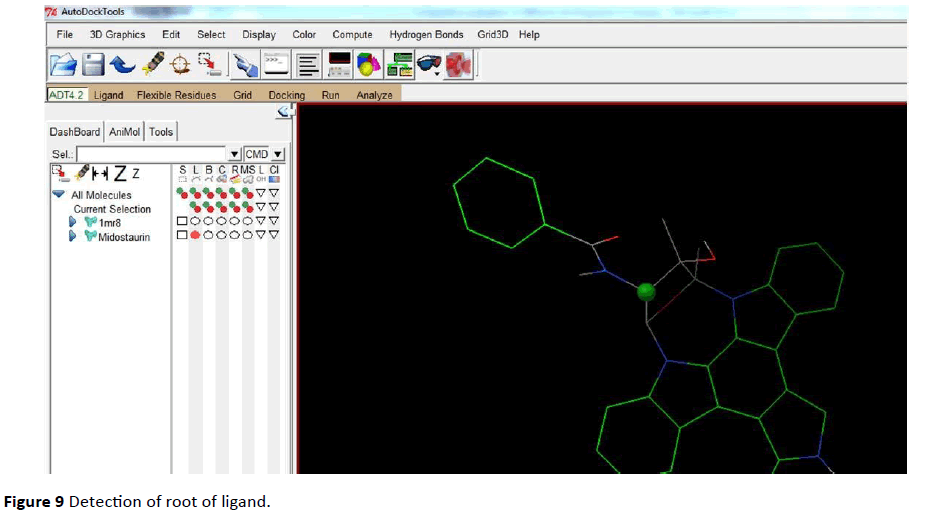
Figure 9 Detection of root of ligand.
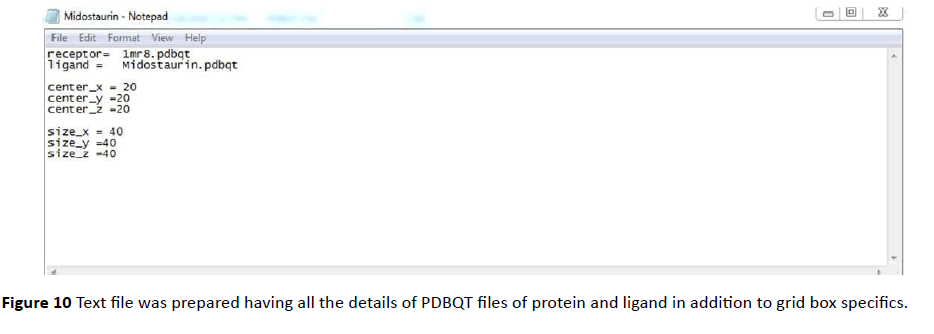
Figure 10 Text file was prepared having all the details of PDBQT files of protein and ligand in addition to grid box specifics.
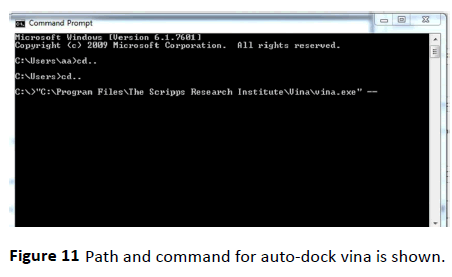
Figure 11 Path and command for auto-dock vina is shown.
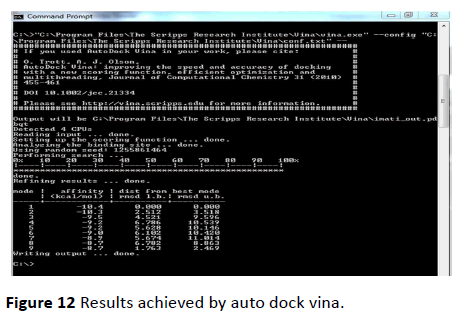
Figure 12 Results achieved by auto dock vina.
Docking with auto dock vina
AutoDock Vina significantly improves the average accuracy of the binding mode predictions.
For its input and output, Vina uses the same PDBQT molecular structure file format used by AutoDock. AutoDock Vina is designed only for receptor-ligand docking. Path and command for auto-dock vina is shown in the below figure (Figure 11).
Docking with Hex server
Both protein and ligand were prepared in PDB format and uploaded in Hex Server. During the process of docking default parameters were used which are already defined and Range angle was selected 180 in addition to step size as 7.5. The number of docking solutions was also not changed and default value of 100 was retained. Output files were downloaded named as “Best Result” in PDB file format.
Docking with patch dock
Protein as well as ligand was uploaded in PDB file format at home page of Patchdock web server. Default parameters were used by retaining 4 A0 for clustering RMSD. Output conformations were generated to 100. All 3 tyrosine kinase complexes were produced by using default parameters.
Results and Discussion
(1MR8) docked with midostaurin
Midostaurin form hydrogen bonds at the required site on receptor molecule at ligand binding site. Glu (69) and Glu(70) in addition to other hydrophobic amino acids like Leu, Val, Ile form ligand cavity. Van der Waals as well as hydrophobic interactions is also involved. Inhibitor constant was calculated as 68.48nM and free binding energy was predicted as -97.77 Kcal/mol (Figures 13-16 and Table 2).
| (1MR8) |
Midostaurin |
Enzastaurin |
| Est. Free Energy of Binding |
-9.77 kcal/mol |
-4.93 kcal/mol |
| Est. Inhibition Constant, Ki |
68.48 nM |
244.28 μM |
| vd + Hbon + desolv Energy |
-7.65 kcal/mol |
-4.66 kcal mol |
| Electrostatic Energy |
-0.03 kcal/mol |
-1.76 kcal/mol |
| Total Intermolecular Energy |
-7.68 kcal/mol |
-6.42 kcal/mol |
Table 2 Binding energies and interaction values of S100A8 dimer docked with inhibitors midostaurin and enzastaurin are shown.
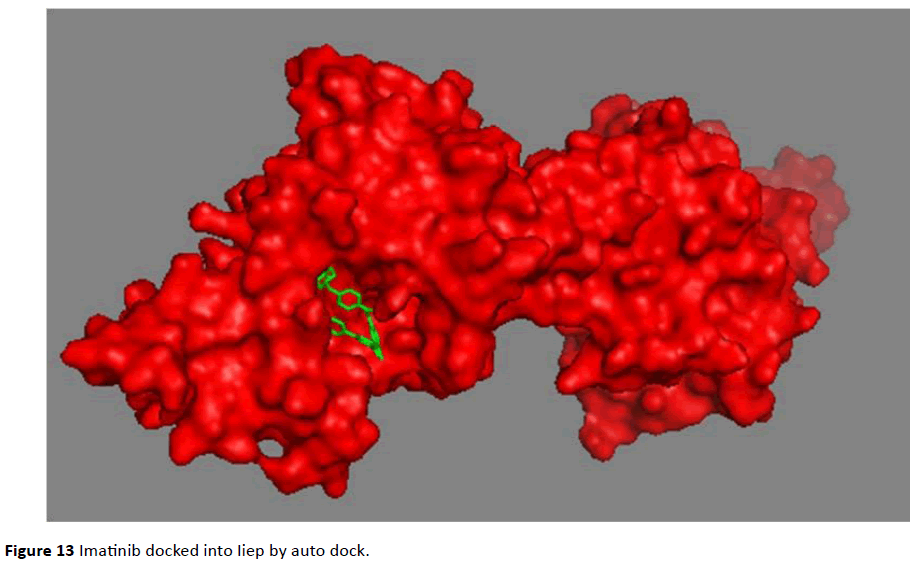
Figure 13 Imatinib docked into Iiep by auto dock.

Figure 14 Imatinib mesylate docked into 1iep results obtained by auto dock vina.
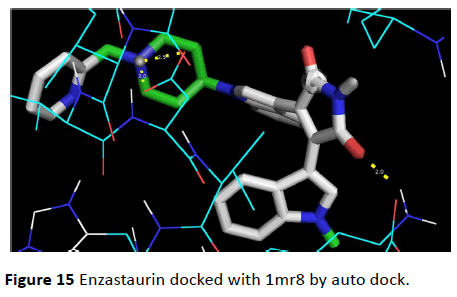
Figure 15 Enzastaurin docked with 1mr8 by auto dock.
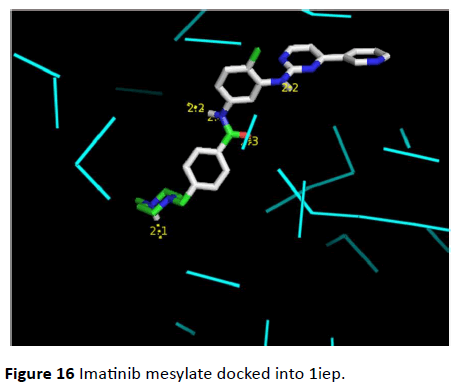
Figure 16 Imatinib mesylate docked into 1iep.
Reference data set description
(1MR8) docked with enzastaurin
This anti-cancerous drug binds at the required site on the receptor molecule forming two hydrogen bonds at ligand binding site. Aliphatic and non-polar amino acids like Lue, Ile and Val form hydrophobic interactions. Inhibitor constant was calculated as 244.28 μM and free energy for binding was calculated as -4.93 Kcal/mol.
Conclusion
Specific study for the purpose of comparative assessment of four docking algorithms has done for the purpose of finding suitable docking tool for kinases. By using Patchdock shape complementarity principles are studied and Hex Server is used to study Fourier transformation correlation. With the help of Auto Dock various molecular force fields methods of proteinligand docking were examined [3]. Dataset for kinases were selected which were used as reference for comparative evaluation of four docking tools which include Auto Dock, Auto dock Vina, Hex Server and Patch dock. In present study Auto dock Vina created the best and most accurate conformation for complexes. This evaluation is very expedient for designing cancer drug against tyrosine kinases.
17665
References
- Cohen P (2002) The origins of protein phosphorylation. Nat Cell Biol 4: 127-130.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912-1934.
- Hubbard SR, Till JH (2000) Protein tyrosine kinase structure and function. Annu Rev Biochem 69: 373-398.
- Paul MK, Mukhopadhyay AK (2004) Tyrosine kinase – Role and significance in Cancer. International Journal of Medical Sciences 1: 101-115.
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: The next generation. Cell 144: 646-674.
- Wang R, Lu Y, Wang S (2003) Comparative evaluation of 11 scoring functions for molecular docking. Journal of Medicinal Chemistry 46: 2287-2303.
- Jiang F, Kim SH (1991) “Soft docking”: Matching of molecular surface cubes. Journal of Molecular Biology 219: 79-102.
- Leach AR (1994) Ligand docking to proteins with discrete side-chain flexibility. Journal of Molecular Biology, 235: 345-356.
- Knegtel RM, Kuntz ID, Oshiro CM (1997) Molecular docking to ensembles of protein structures. J Mol Biol 266: 424-440.
- Lorber DM, Shoichet BK (2005) Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem 5: 739-749.
- McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK (2003) Gaussian docking functions. Biopolymers 68: 76-90.
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M (2003) Ligand Fit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 21: 289-307.
- Ewing TJ, Kuntz ID (1997) Critical evaluation of search algorithms for automated molecular docking and database screening. Journal of Computational Chemistry 18: 1175-1189.
- Rarey M, Kramer B, Lengauer T, Klebe G (1996) A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261: 470-489.
- Grieshammer S, Grope OH, Koettgen J, Martin M (2014) A combined DFT + U and Monte Carlo study on rare earth doped ceria. Physical Chemistry Chemical Physics 16: 9974-9986.
- Sousa SF, Fernandes PA, Ramos MJ (2006) Protein–ligand docking: Current status and future challenges. Proteins: Structure, Function, and Bioinformatics 65: 15-26.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267: 727-748.
- Moreland L, Bate G, Kirkpatrick P (2006) Abatacept. Nat Rev Drug Discov 5: 185-186.
- Li X, Li Y, Cheng T, Liu Z, Wang R (2010) Evaluation of the performance of four molecular docking programs on a diverse set of protein-ligand complexes. Journal of Computational Chemistry 31: 2109-2125.
- Kontoyianni M, McClellan LM, Sokol GS (2004) Evaluation of docking performance: comparative data on docking algorithms. J Med Chem 47: 558-565.
- Baxter CA, Murray CW, Clark DE, Westhead DR, Eldridge MD (1998) Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins 33: 367-382.
- Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP (1997) Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des 11: 425-445.
- Mooij WT, Verdonk ML (2005) General and targeted statistical potentials for protein-ligand interactions. Proteins 61: 272-287.
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, et al. (2016) Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protocols 11: 905-919.
- Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW (2010) Hex Server: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Research 38: 445-449.
- Jain AN (2003) Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem 46: 499-511.
- Ruppert J, Welch W, Jain AN (1997) Automatic identification and representation of protein binding sites for molecular docking. Protein Sci 6: 524-533.
- Jain AN (1996) Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des 10: 427-440.
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) Patch Dock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Research 33: 363-367.
- Kellenberger E, Rodrigo J, Muller P, Rognan D (2004) Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 57: 225-242.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. (2000) The Protein Data Bank. Nucleic Acids Research 28: 235-242.