Introduction
In cattle a lethal genetic disease called Bovine Leukocyte Adhesion Deficiency (BLAD) has been reported that affect the hemopoeatic system of an animal. In the last decade, this disease gained significant economic importance. It has also been recorded in canine and human population (GERARDI, 1996).
The molecular basis of this disease involves an aspartic acid to glycin substitution at amino acid 128 of the gene encoding CD18 sub-unit of beta 2 integrin. The mutation is considered lethal because homozygous animal dies before reaching sexual maturity. In 1990s, this disease was the most common and widespread genetic disorder in HF cattle worldwide (CZARNIK et al., 2007). For example, the occurrence BLAD heterozygous sires and heifers during the period 1993-1998 in Czech Holstein was 13.9% and 10.7 %, respectively (CITEK et al., 2008).
India embraced artificial insemination as the method of rapid dissemination of superior genetic potential within indigenous cattle breed in the country. During the period between 1961 and 1978, it had imported around 1470 bulls and 6165 females of exotic breeds of which 275 bulls and 1825 females were of Friesian and HF breeds while most others were Jersey. More than 23 farms of HF breed together contributed to the semen freezing network majority of which were established under the Operation Flood Program in cooperation with National Dairy Development Board with assistance from Governments of Switzerland, UK and USA (MUKHOPADHYAYA AND MEHTA, 2002).
This indicates that a significant inflow of exotic genetic material within the country had occurred that formed the basis of enhancement in heritable productivity enhancement capabilities within the organized diary sector in the country. BLAD was first identified in North American Holsteins which were routinely exported to different other national cattle populations in the world (KERHLI et al., 1990).
The molecular basis of this disease is interesting. It occurs due to deficient CD18 proteins generated due to single nucleotide polymorphism in the corresponding gene. These proteins are component of adhesion molecules on the surface of neutrophils. Adhesion of neutrophils to the endothelial cells is an important step in immediate combating of infections. Due to this mutation, the neutrophils fail to adhere to the endothelial cells this compromising with the immediate immune response capability of an individual when challenged by a pathogen. Hence animals homozygous for this mutation suffer from recurrent infection and require continued antibiotic therapy (HEALY, 1996). It is because of this, most descendents of North American Holsteins including the well known Ivanhoe Bell, a BLAD founder bull, replaced those of LMKK in Australian artificial breeding centres (HEALY, 1996).
Several other countries including Japan (NAGAHATA et al., 1997), Germany (TAMMEN et al., 1996) and India (MUKHOPADHYAYA AND MEHTA, 2005) have taken proactive steps to eradicate this genetic disorder from their breeding population (MUKHOPADHYAYA et al., 2000).
There are several methods developed for detection of the ‘A’ to ‘G’ mutation in the CD18 gene. These include non-isotopic Ligase Chain Reaction (BATT et al., 1994) and PCR-RFLP (SHUSTER et al., 1992). However, the most common method of detection of this genetic disease is the one based on PCR-RFLP because of the cost effectiveness and ease of operation.
There are several variants of PCR-RFLP based detection method for BLAD in cattle. One of them, is setting up of a multiplex PCR reaction where a region containing the mutational hotspot within the CD18 gene as well as that of k casein gene is co -amplified and subjected to RFLP analysis to predict the carrier status of an animal as well as identify the genotype for the milk production trait gene (ZSOLNAI AND FESUS, 1996). Nevertheless, the most common one is amplification of a 58 bp fragment harboring the D128G (A‡G) mutation followed RFLP analysis using Taq I restriction endonuclease. The normal allele gets restricted into 25 bp and 33 bp respectively, while the mutant allele remains redundant to restriction digestion (MUKHOPADHYAYA et al., 2000). It has been observed that detection of 58, 25 and 33 bp fragments after routine resolution on an agarose gel is inconvenient when large numbers of samples are to be screened owing to the small difference as well as size of the fragments. This inconvenience is amplified when non-denaturing polyacrylamide gel is used due to the intricacies involved in casting, running and handling non denaturing polyacrylamide gels.
In this study, we report a new set of oligonucleotide primers that is suitable for amplifying a fragment larger than 58 bp which after restriction digestion with Taq I generates a profile that is easier to analyze based on differential migration of one of the restricted fragments on a regular agarose gel electrophoresis. This modification of the original protocol published by SHUSTER et al., (1992) adds convenience and speed to large scale, blind screening programme for detecting carrier for BLAD in organized dairy sector in India.
Materials and methods
Sample collection and processing
Panel A comprised of blood samples collected in EDTA vacutainer tubes (BD, USA) from male animals (n=75) of undisclosed breeds reared at different villages and farms at Surat, Gujarat (India) under Surat Milk Union Limited (SUMUL) and shipped to geneOmbio Technologies central processing laboratory at room temperature within 48 hours from the time of collection. The genomic DNA was isolated and assessed for purity following standard molecular biology protocols (SAMBROOK AND RUSSELS, 2001).
Panel B comprised of archived genomic DNA isolated from venous blood of different breeds of cattle (Holstein Friesian=156; Jersey=105) purified by the phenol chloroform method (SAMBROOK AND RUSSELS, 2001) and stored as DNA pellet suspended in 70% alcohol at -200C. Prior to use, the DNA solution was centrifuged at 14000 rpm for 15 minutes, supernatant discarded, pellet dried at room temperature, suspended in sterile water, heated to 450C for 10 minutes and then used for PCR. The quality and quantity of DNA samples were estimated by agarose gel electrophoresis (0.8%) using standard protocol (SAMBROOK AND RUSSELS, 2001) and spectrophotometric reading at 260 nm.
Designing of de novo oligonucleotide primers
The nucleotide sequence of CD18 gene of cattle was accessed from the public domain (https://www.ncbi.nlm.nih.gov) (Gene bank accession number Y12672). The hotspot for mutation at amino acid position 128 (nucleotide position 383) was subsequently identified and oligonucleotide primers designed bracketing the SNP using the Primer 3 software (STEVE ROZEN et al., 2000).
In silico RFLP analysis
The nucleotide sequence of the fragment targeted for amplification by the new primer sets (this study) to restricted in silico with Taq I and the migration vis a vis uncut PCR amplicon was checked using an online software (NEW ENGLAND BIOLAB, USA). In silico restrictions mapping of the fragment with other enzymes flanking the unique Taq I were also done using the same software.
PCR and RFLP analysis
Oligonucleotide primers were synthesized by the phosphoramidate method from Sigma Aldrich (India), Bangalore. For amplifying the 184 bp fragment, approximately 100 ng of DNA were used for PCR amplification using an automated thermal cycler (GeneAmp PCR System, model 9700; Applied Biosystems, Foster City, Calif, USA). The PCR mix contained 2.5 _l of 10x PCR buffer (100 mM Tris – pH 9.0, 500 mM KCl, 15mM MgCl2 and 0.1% Gelatin), 200 _M dNTPs, 1 unit of Taq DNA polymerase (Vivantis, Malaysia), 20 pMoles each of OF primer (5’- GTCAGGCAGTTGCGTTCA - 3’) and OR primer (5’- GTAGCTGCCTCACCAATG - 3’) and sterile nucleasefree water to make a final volume of 25 _l. The samples were amplified at two stages. Stage one included 20 cycles, each cycle comprising of 940C hold for 30 seconds (denaturation) followed by 700C for 20 second (primer annealing) and 720C for 20 second (primer extension). This was followed by stage two that included another 15 cycles, each comprising of 940C hold for 30 seconds (denaturation), 650C for 20 seconds (primer annealing) and 720C of 20 second (primer extension). Fifteen _l of the PCR product was digested with 5 units of Taq I restriction endonuclease for 3 hours at 650C and analyzed by electrophoresis (3% agarose, SRL laboratories, India) spiked with ethidium bromide (0.5 _g/ml) in 0.5% TBE buffer (SAMBROOK AND RUSSELS, 2001) and visualized under UV transilluminator (260 nm.) Amplification of the 58 bp region of CD18 gene containing the mutation hotspot was performed using published protocol (SHUSTER et al., 1992).
Results and discussion
The occurrence of BLAD is due to a single nucleotide polymorphism (SNP) within the exonic region of the CD18 gene. Since RFLP is a robust technique for identification of SNP particularly in heterozygote condition, in this study this feature was preserved for detection of genetic mutation leading to BLAD but the size of the PCR amplicon was increased to facilitate superior resolution of fragments and easier detection.
Our strategy was to avoid inclusion of additional Taq I recognition sites within the PCR amplicon other than the diagnostic one constituting codon 128 to simplify the process of detection. Yet another objective was not to increase the size of the PCR amplicon beyond 200 bases to ensure efficient PCR reaction. We found that there was a single Taq I site 540 bp bases upstream and 447 bases downstream of the one that included the diagnostic SNP (A‡G) constituting codon 128 of the CD18 gene. This led us to design the forward and reverse primers such that their 5’ ends were spaced 51 and 129 bases away from 5’ and 3’ end of this diagnostic Taq I site respectively, so as to get the most optimum set of PCR primers (Figure 1).

Figure 1: Primer binding sites on the CD18 gene of cattle. Shaded arrows with underlined nucleotide sequences are primers designed in this study; plain black arrows with nucleotide sequence in italics are published primers (SHUSTER et al., 1992). The diagnostic SNP is shown in a black box and the position (in nucleotides from the start codon of the gene) with a black vertical arrow.
These primers were expected to generate PCR amplicon of size 184 bases that upon restriction by Taq I would cleave into two fragments of sizes 52 and 132 bases, respectively.
The nucleotide sequence targeted for amplification using OF and OR primers was subjected to in silico restriction enzyme mapping (Figure 2). It was found that the Taq I site was unique and asymmetrically placed within this fragment. This was further confirmed by running an in silico RFLP experiment to generate a profile that was anticipated after actual experimentation (Figure 3). In all cases, for comparison, parallel analysis was done for the 58 bp sequence also (SHUSTER et al., 1992).
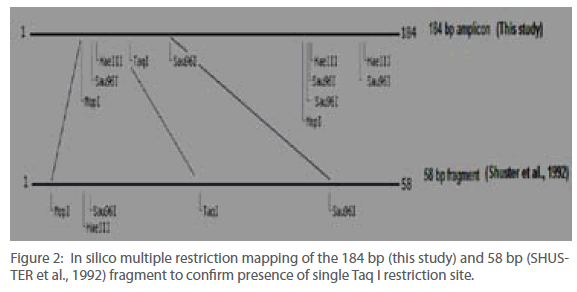
Figure 2: In silico multiple restriction mapping of the 184 bp (this study) and 58 bp (SHUSTER et al., 1992) fragment to confirm presence of single Taq I restriction site.

Figure 3: In silico RFLP profile of the 184 (A) and 58 bp (B) PCR fragment using Taq I restriction endonuclease (3% agarose). M: DNA size standard; lane 1: undigested PCR fragment; 2: PCR fragment digested with Taq I and representing an affected animal; 3: Digested PCR fragment mixed with undigested one to simulate a carrier RFLP profile. Sizes of the fragments are shown in base pairs (bp).
The data thus generated was in line with expected results and therefore supported actual experimentation on cattle DNA for testing occurrence of BLAD either in carrier or affected form.
For this, we used two panels of resource population. Panel A comprised of cattle DNA from undisclosed breeds collected from different farms at Gujarat (India) while panel B comprised of descriptive breeds of cattle comprising of Jersey and HF Crossbreds. A total of 336 animals were assayed for this study. The BLAD testing protocol published by SHUSTER et al., (1992) was considered as the gold standard.
PCR amplification of purified DNA using OF and PR primer pair generated a 184 bp fragment (Figure 4, lane no. 1) as anticipated. This PCR product upon restriction digestion with Taq I generated 132 and 52 bp fragments, respectively (Figure 4, lane no. 2 & 3 respectively). In order to simulate the RFLP profile of carrier animal, uncut PCR amplicon was added to that restricted with Taq I in equimolar ratio prior to resolving the fragments on an agarose gel. This lane therefore contained three fragments sized 184, 52 and 132 bp, respectively (Figure 4, lane no. 4). For the screening programme, every agarose gel run comprised of, apart from test samples, an uncut PCR amplicon, DNA size standard and simulated carrier for comparison.
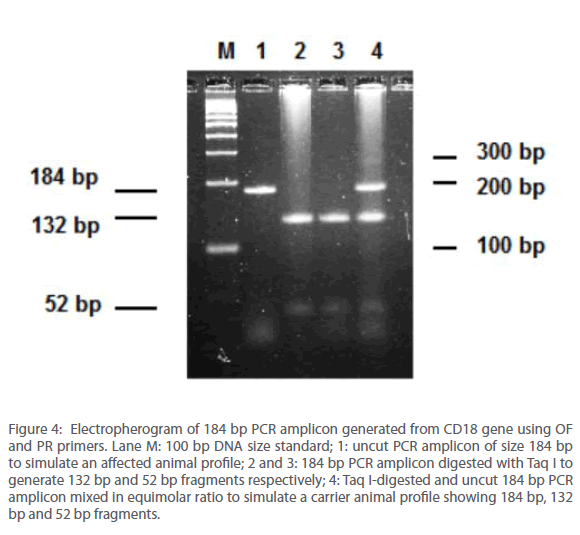
Figure 4: Electropherogram of 184 bp PCR amplicon generated from CD18 gene using OF and PR primers. Lane M: 100 bp DNA size standard; 1: uncut PCR amplicon of size 184 bp to simulate an affected animal profile; 2 and 3: 184 bp PCR amplicon digested with Taq I to generate 132 bp and 52 bp fragments respectively; 4: Taq I-digested and uncut 184 bp PCR amplicon mixed in equimolar ratio to simulate a carrier animal profile showing 184 bp, 132 bp and 52 bp fragments.
It was observed that upon appropriate resolution on a 3% agarose gel run for 40 minutes using a 0.5X TBE buffer (SAMBROOK AND RUSSELS, 2001), the migration difference of the 184 bp and 132 bp fragments were sufficient to diagnose presence or absence of SNP at codon 128. The 52 bp fragment was visible in all cases (Figure 4, lane no. 2, 3 and 4 respectively) but was not found essential for diagnosis. On the other hand, detection of a 58 bp as well as at least one additional fragment of size 33 bp fragment (and preferably another of size 25 bp) was found essential for concluding an assay using the gold standard.
Polyacrylamide gel electrophoresis is the method of choice for resolving fragments smaller than 50 bp (RAPLEY, 2000). However, this method is cumbersome and involves handling of the fragile polyacrylamide gels. On the other hand, there is specialized agarose commercially available (Cambrex Bio Science Rockland, Inc.) for resolving small sized PCR or restricted DNA fragments but are expensive compared to regular ones (e.g., SRL, India) available in the market and therefore not economical for routine use in large scale. Also, there are reports of improvised methods for formulating gelling medium for resolving small DNA fragments (PERLMAN et al., 1987) but the components are not available commercially and involve less convenient formulation steps.
Since the first report of occurrence of BLAD in cattle population in India by our group (MURALEEDHARAN et al., 1999), we were also the first to update frequency of prevalence of this genetic disease (0.42%) and other similar disorders in India through systematic reporting in scientific journal (MUKHOPADHYAYA AND MEHTA, 2005). This trend was followed by PATEL et al., (2007) where it was reported that the percentage of BLAD carriers in HF and HF crossbred population in India increased to 3.23%. This indicates a sharp rise in frequency of occurrence of carriers in organized dairy sector in the country indicating a situation that is alarming. The observation underlines the need for a large scale screening of cattle in the organized dairy sector within India for this genetic disorder in order to identify carriers especially for those farms that maintain exotic crossbred cattle population.
Results obtained from screening exercise of the resource population using the published (SHUSTER et al., 1992) (Figure 5) and newly designed (this study) primers (Figure 4) indicated that the resolution and clarity of detection is significantly improved in the test with newly designed primers. An important aspect of this new assay was generation of the 52 bp fragment that originated from a normal allele, after restriction digestion. It was formed due to asymmetric position of the Taq I site on the 184 bp PCR amplicon. However, since the profile and migration difference between the larger fragments, viz., 184 bp and 132 bp was significant and unambiguous, they were found to be sufficient for diagnosis of BLAD alleles and detection of 52 bp restricted fragment that originate from a normal allele was not required (Figure 4). Further, this resolution could also be done using any regular agarose (SRL, India) with low endosmosis value and did not call for expensive and exclusive variants of any gelling medium specialized for detection and resolution of small DNA fragments.
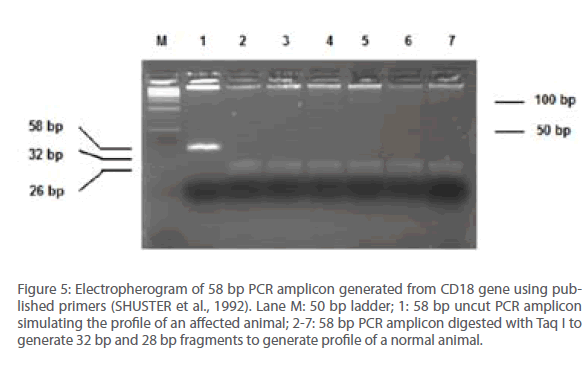
Figure 5: Electropherogram of 58 bp PCR amplicon generated from CD18 gene using published primers (SHUSTER et al., 1992). Lane M: 50 bp ladder; 1: 58 bp uncut PCR amplicon simulating the profile of an affected animal; 2-7: 58 bp PCR amplicon digested with Taq I to generate 32 bp and 28 bp fragments to generate profile of a normal animal.
MUKHOPADHYAYA et al., (2000) developed the MIS technology for developing synthetic controls to facilitate uninterrupted supply of carrier and affected control DNA for large scale screening of cattle and buffalo population for BLAD. These control-DNA were generated through a modified site-directed mutagenesis protocol that relied on incorporation of a mutation using one of the PCR primers as the vehicle. However due to spacing of the diagnostic SNP by several nucleotides from both forward and reverse primers to generate the 184 bp amplicon, the MIS method cannot be used to generate similar synthetic controls for the assay reported in this study.
We concluded that the de novo primers designed in this study can be utilized as a reliable substitute for the published primers by SHUSTER et al., (1992) and can also be used with greater convenience using lesser percent of regular agarose and superior resolution of fragments for detection of cattle carrier for BLAD in organised dairy sectors in India and other countries. Blind screening of cattle is one of the most effective methods of controlling the incidence of such genetic disorders. Therefore improved methods of detection for this disease will go a long way in facilitating and encouraging large scale screening of cattle in smaller laboratories with infrastructure limitations for this economically important disease in India and other affected countries facing similar conditions where artificial insemination is in vogue and is being used rampantly for horizontal transmission of genes within the dairy animal population as in India (MUKHOPADHYAYA AND MEHTA, 2002).
Genetic disorders such as BLAD do not have effective medicine to combat and further, the mortality rates are high. This therefore warrants the need for a continuously evolving system of diagnostics with strong focus on efficacy, convenience and economy. The present study addressed this need and establishes an improved protocol for detection of an economically important genetic disorder in cattle which has potential for use in other mammals also with special reference to humans.
Acknowledgement
Collection of blood samples that comprised Panel A in this study was facilitated by PM Patel and his team and processed by Umakant M and Suresh B at geneOmbio Technologies central processing laboratory. Data was compiled and archived by Mrinal Virkar. Assistance of all staff at MBPS College, Anand is acknowledged for storage of Panel B samples used in this study. Encouragement and support from Sachin Purohit, Bikash Aich, Rahul Bavadekar and P R Pandey for conducting and concluding this study is thankfully acknowledged.
383
References
- Batt C A, Wagner P, Wiedmann M, Luo J, Gilbert R (1994). Detection of bovine leukocyte adhesion deficiency by nonisotopic ligase chain reaction. Anim Genet 25:95-98.
- Cítek J, Rehout V, Schröffelová D, Hradecká E (2008). Frequency of BLAD and CVM alleles in sires and elite heifers of Czech Holstein cattle. Dtsch Tierarztl Wochenschr 115:475-477.
- Czarnik U, Grzybowski G, Kaminski S, Prusak B, Zabolewicz T (2007). Effectiveness of a program aimed at the elimination of BLAD-carrier bulls from Polish Holstein-Friesian cattle. J Appl Genet 48:375-377.
- Gerardi A S (1996). Bovine leukocyte adhesion deficiency: a review of a modern disease and its implications. Res Vet Sci 61: 183-186.
- Healy, P J (1996). Testing for undesirable traits in cattle: an Australian perspective J Anim Sci 74:917-922.
- Kerhli, M E., Schmalstieg, F C, Anderson, D C, Van Der Maaten, M J, Hughes, B J, Ackermann, M R, Wilhelmsen, C L, Brown, G B, Stevens, M G, and Stone, C A (1990). Molecular definition of the bovine granulocytopathy syndrome: Identification of deficiency of the Mac-1 (CD11b/CD18) glycoprotein. Am. J. Vet. Res 51:1826-1830.
- Mukhopadhyaya P N and Mehta H H (2001). Genotype profiles for the quantitative trait related to milk composition in bulls used for artificial insemination in India. Asian Aust J Anim Sci 15:326-329
- Mukhopadhyaya P N and Mehta H H (2002). Identifying an alternative method of blood collection, transportation and storage for routine PCR-based diagnostic tests in cattle. Ind Vet J 79:887-890
- Mukhopadhyaya P N and Mehta H H (2005). Incidence report update of three economically important genetic diseases in breeding bulls of organized dairy sectors. Ind Vet J 82: 1015-1016
- Mukhopadhyaya P N, Mehta H H and Rathod R N (2000). A method for the simulation of normal, carrier and affected controls for PCR-RFLP screening of genetic disease in dairy cattle. Molecular and Cellular Probes 14:381-384
- Muraleedharan P, Khoda V K, Mukhopadhyaya P N, Mehta H H and Schwerin M (1999). Incidence of hereditary Citrullinemia and Bovine Leukocyte Adhesion Deficiency Syndrome in Indian Dairy Cattle and Buffalo population. Arch Anim Breeding 42: 347-352
- Nagahata H, Masuyama A, Masue M, Yuki M, Higuchi H, Ohtsuka H, Kurosawa T, Sato H, Noda H (1997). Leukocyte emigration in normal calves and calves with leukocyte adhesion deficiency. J Vet Med Sci 59:1143-7.
- Perlman, D, Chikarmane, H and Halvorson, HO (1987). Improved resolution of DNA fragments in polysaccharide-supplemented agarose gels. Analytical Biochemistry 1: 247-254.
- Rapley, R (Edited) The Nucleic Acid Protocols. Handbook, Humana Press, Tolowa, NJ (2000).
- Sambrook J and Russell D W (2001). Molecular Cloning: A Laboratory Manual Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Shuster D E, Kehrli M E Jr, Ackermann M R, Gilbert R O (1992). Identification and prevalence of a genetic defect that causes leukocyte adhesion deficiency in Holstein cattle. Proc Natl Acad Sci USA 89:9225-9229.
- Rozen S and Skaletsky H J (2000). Primer 3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols: Methods in Molecular Biology, Humana Press, Totowa, NJ, 365-386.
- Tammen I, Klippert H, Kuczka A, Treviranus A, Pohlenz J, Stöber M, Simon D, Harlizius B (1996). An improved DNA test for bovine leucocyte adhesion deficiency. Res Vet Sci 60:218-221.
- Zsolnai A, Fésüs L (1996). Simultaneous analysis of bovine kappa-casein and BLAD alleles by multiplex PCR followed by parallel digestion with two restriction enzymes. Anim Genet 27:207-209.


