Introduction
Rheumatic diseases cause a high demand on health services, it has been estimated that about 33% of the general population at some point in their lives has a rheumatic disease [1].
RA is one of the three most common diseases in the outpatient Rheumatology with an estimated prevalence of up to 47.1% [2].
This disease presents a very real risk of functional impairment, since it was found that a decade after the onset of symptoms, at least 50% of patients are unable to maintain a full time job [3].
Additionally, patients with RA have an increased in the mortality rate [4].
The etiology of RA remains unknown. Like other autoimmune diseases, is characterized by an alteration in the immune response in the presence of chronic inflammation and production of autoantibodies (Figure 1).
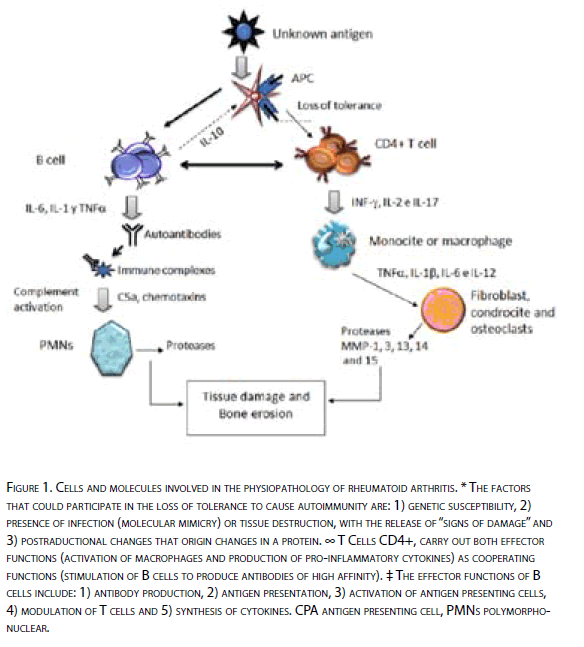
Figure 1: Cells and molecules involved in the physiopathology of rheumatoi d arthritis. * The factors that could participate in the loss of tolerance to cause autoimmuni ty are: 1) genetic susceptibility, 2) presence of infection (molecular mimi cry) or tissue destruction, wi th the release of “signs of damage” and 3) postraductional changes that origin changes in a protein. ∞ T Cells CD4+, carry out both eff ector functions (activation of macrophages and production of pro-i nflammatory cytokines) as cooperating functions (stimulation of B cells to produce antibodies of high aff inity). ‡ The eff ector functions of B cells include: 1) antibody production, 2) antigen presentation, 3) activation of antigen presenting cells, 4) modulation of T cells and 5) synthesis of cytokines. CPA antigen presenting cell, PMN s polymorphonuclear.
Through the years, several paradigms have been broken in what respects to the pathogenesis of this disease. Weyand and Goronzy in 1997 [5] proposed a new model for the physiopathology of RA, which integrates genetic risk factors and the complexity of the inflammatory responses.
This model assumes that the host’s immune response is not involved in the initial process of the disease, but in a first stage, the host is exposed to a large number of antigens secondary to synovial tissue damage, by multiple causes and that in parallel, several genes of the immune response such as HLA, immunoglobulin genes and T-cell receptors, have an impact on the kind of response that develops against these antigens. In a second stage, then, arises the autoimmune response, where the immune system cells, specifically self-reactive T cells, create a response against these antigens and origins an inflammatory process around, with the presence of cells such as macrophages, T cells, B and neutrophil embedded in the inflamed synovial membrane. The production of antibodies and immune complex formations, which in turn causes the recruitment of a greater amount of inflammatory cells and their products [6].
During the last decade, research on the pathogenesis of RA has focused on the idea that there is involvement of multiple effector cells and molecular mechanisms of the immune system. However, from this complex view emerges a common point, that is, to escape the immune control. In this way the fundamental aspects in the pathogenesis of the disease are the understanding of the loss of tolerance, the activation of autoreactive T cells and the generation of autoantibodies.
HLA: The shared epitope
Several studies have already shown a strong correlation between the presence of RA and some alleles of HLA-DR. The HLA-DRB1 * 0401 and HLA-DRB1 * 0404 (encoding the molecule DR4dw4) are most commonly found and their presence is associated with a more severe disease with high mortality [7,8]. These HLA-DR molecules share a common sequence in the third hyper variable region (shared epitope), these reasons consist of amino acids 70QRRAA74 in 70QKRAA74 in DRB1 * 0401 and DRB1 * 0404, respectively [9]. These five amino acids are found at the peptide union point and are therefore of utmost importance during the antigen presentation.
Genetic studies, which assess the contribution of HLA to the physiopathology of RA, have shown that the risk to suffer from this disease in siblings of affected individuals is 2-17 times higher than in unaffected siblings of individuals [10]. It has also described the association between RA and the HLA complex in different populations, being up to approximately one third of genetic component for susceptibility in the disease [10].
Autoantibodies in the Physiopathology of RA
A prominent feature of RA is the presence of several types of autoantibodies. Furthermore, this disease is one of the few in which the presence of structures similar to ectopic germinal centres can be found in the inflamed synovial membrane [11,12]. This feature suggests the presence, activation, differentiation and local production of antibodies by B cells [13].
The contribution of these antibodies to the disease starts with direct union of these antibodies to their antigens, immune complex formation, deposition and complement activation and Fc receptors, production of chemiotactic factors and recruitment of polymorphonuclear involved in the inflammatory reaction by the production of proteases that cause tissue damage [14,15].
The antibody best known and used is the rheumatoid factor (RF), which is an autoantibody directed against the Fc portion of IgG molecules. Although it is not specific to the disease is routinely used for the classification of RA [16].
Recently emerged interest in a new class of autoantibodies specific for RA, antibodies against cyclic citrullinated peptide (aPCC), which achieve a 98% of specificity and sensitivity of 80% [17,18]. These antibodies aPCC belong to the family of autoantibodies against perinuclear factor (APF) [18,19]. The epitope of this group of antibodies are the waste in which the arginine is converted by the peptidyl arginine desaminase (PAD) to a citrullinated product through postraductional modifications [20,21]. From there, this group of autoantibodies are known as antibodies against citrullinated proteins.
One study in particular, highlights the presence of these antibodies in asymptomatic individuals (blood donors) who subsequently would develop RA; in this way, they are given a high positive predictive value [22]. This indicates that the citrullination of proteins and antibodies production, are early processes involved in the development of AR [23,24].
On the other hand, the presence of citrullinated proteins in sinovium is not associated to the presence of aPCC in patients with AR, since it was demonstrated that even when there are citrullinated proteins in inflamed sinovium because of different factors, other than RA, it is not detected the presence of antibodies aPCC [25]. For this reason, it has been postulated that the presence of these proteins with citrullinated peptides may be a phenomenon associated with inflammation [26], this would explain the presence of citrullination in inflammatory arthropathies different to RA.
In a study by Verport et al [27], it was demonstrated the presence of aPCC IgM class, on a continuous basis during the course of RA, this indicates that during the disease there is an active recruitment of new B cells to the immune response originally mounted, reflecting an ongoing re-activation and persistence of the trigger antigen of the production of antibodies against citrullinated residues.
There are two possible explanations for the involvement of these antibodies in the pathogenesis of RA:
The first is that, secondary to an increase in the amount of citrullinated antigens, specific of RA, there is a specific immune response and thus the production of specific antibodies. However, the presence of citrullinated proteins has proven to be a phenomenon present in any inflamed synovial tissue [25]. The second possibility is that patients with RA may have an abnormal humoral response to these proteins and produce in an exaggerated form a large amount of antibodies.
Recently it was demonstrated the presence of antibodies directed against the enzyme PAD IV in serum of patients with RA [28], both in Japanese and Caucasian populations. Although rare, its presence is highly specific when compared with Systemic Lupus Erythematosus SLE and healthy subjects. In addition, we were able to establish an association between high concentrations of these antibodies with a more severe AR and it was strongly associated with the presence of antibodies aPCC. The formation of complexes between PAD IV and its substrate (citrullinated peptide) could act as hapten carrier and be recognized by B cells through BCR either specific for PAD IV or citrullinated substrate. In this way, it is possible the production of antibodies against either the enzyme PAD IV or against the citrullinated peptide and present any epitope to a specific CD4+ T cell.
Citrullinated Residues
Under normal conditions only few proteins contain citrullinated residues. Citrullinated residues, must differentiate from citrulline in a free form, whose metabolism begins in the small intestine, where it synthesizes and ends in the kidney, where it is degraded [29]. This citrulline is produced regardless of citrulline in a peptide form. The generation of citrulline in a peptide form is dependent of PAD enzymes, which as part of postraductional modifications, convert basic arginine residues to citrulline residues neutral. To date we have identified five isotypes of this enzyme. Enzymes are distributed in different tissues: PAD I is mainly expressed in epidermis and in the female reproductive tract; PAD II in skeletal muscle, spleen, brain and secretory glands; PAD III in pilose follicles, PAD IV in neutrophils, eosinophils and skeletal muscle; PADVI in ovaries, embryos in early stages of development and zygotes [30].
There is a theory that supports the possibility of an increase in the amount of citrullinated antigens, this suggests that the presence of polymorphisms and even the presence of a particular haplotype was associated with the presence of RA, as described by Suzuki et al [31] . It consists of four single nucleotide polymorphisms (SNPs) located in exons 2, 3 and 4 of the gene PADI4, located on chromosome 1, region 1p36, which encodes for the enzyme peptidyl arginine desaminase IV (DAP IV). The presence of this susceptibility haplotype in Japanese population was associated with an increased stability of the transcript [31,32] Indeed, in theory, could lead to an increase in levels of PADIV and hence the citrullination of more epitopes. These citrullinated epitopes then become a target for auto-antibodies specific to RA (aPCC antibodies) and when a greater number of these immune complexes is generated would favor the generation of this autoimmune disease. This theory is verified in part after Vossenaar et al findings [33], when reporting that patients who were homozygous for the susceptibility haplotype had significantly higher degrees of aPCC antibodies (87% vs. 67%, p <0.05 ) compared with patients heterozygous and homozygous for the non-susceptibility haplotype. However, it is still necessary to verify that the enzyme levels are high, and even high levels of citrullinated antigens in individuals carrying the susceptibility haplotype.
It is also important to note that not only the presence of cells expressing these enzymes in synovial tissue is a prerequisite for the development of citrullinated antigens, as for the activation of those enzymes normally located intracellularly, an increase in concentration of cytosolic Ca+2 is necessary [34].
During cell death, the integrity of the plasma membrane is lost, the enzymes can then leave the cell and activate as the concentrations of extracellular Ca +2 exceed the threshold of activation of those enzymes (10-5 mM) in this way, it is induced the citrullination of extracellular proteins or can be activated inside the cell to increase the influx of Ca+2 and in this way citrulline intracellular proteins [35].
Moreover, given the conversion of arginine to citrulline, the amino group of the positively charged lateral chain is changed to a neutral carboxyl group, which would affect the ability of peptides to bind covalently with HLA molecules.
In DRB1 * 0401 transgenic mice, the citrullination of peptides not only increased the affinity between peptide and the major histocompatibility complex (MHC) but also led to activation of CD4+ T cells [36].
Moreover, this catalytic process, can cause changes in the primary, secondary and tertiary protein structure (Figure 2). In vitro studies have shown that high citrullination can cause protein denaturation [37]
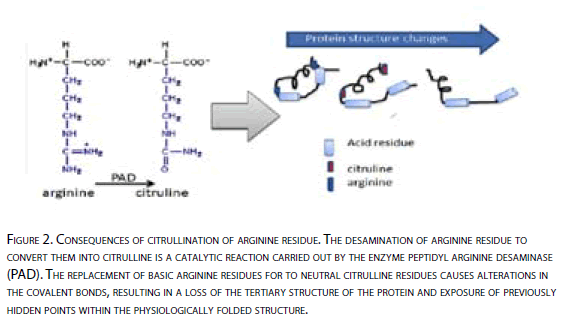
Figure 2: Consequences of citrullination of arginine residue. The desamination of arginine residue to convert them into citrulline is a catalytic reaction carried out by the enzyme peptidyl arginine desaminase (PAD ). The replacement of basic arginine residues for to neutral citrulline residues causes alterations in the covalent bonds, resulting in a loss of the tertiary structure of the protein and exposure of previously hidden points within the physiologically folded structure.
However, why as a physiological process as is citrullination, would be firing autoimmune responses?
Possible explanations for which the generation of citrulline in peptide form, being a physiological process and present in most individuals is soon recognized as strange or dangerous could be: 1) increase in production, resulting in loss of tolerance T cells, 2) impairment in the ability of these peptides to bind to molecules in the presence of the HLA shared epitope and 3) change in three-dimensional structure of the protein resulting in exposure of primary or secondary structures, not being exposed before to the immune system, or alterations in the binding of these proteins with others.
Escape To the Mechanisms of Tolerance
The immune system has evolved in an efficient way to remove what it does not belong to it or what it is dangerous according to Matzingen [38]. The establishment and maintenance of tolerance towards what is part of it is a fundamental property of the immune system, and fails in this tolerance leads to the generation of autoimmunity.
Central tolerance involves the elimination of auto-reactive T cells during their development in the thymus, since the only class of antigens present in high concentrations in this body, are own antigens. In this way, those lymphocytes that recognize and react against own antigens are eliminated (negative selection). However, a number of auto-reactive T cells escape to this selection and exit to the periphery. These lymphocytes could activate and cause autoimmunity. Peripheral tolerance ensures that self-reactive T cells that escaped the control of central tolerance are eliminated, being leaded to the apoptosis or are unable to mount an immune response remaining in anergic state. Foreign antigens, not dangerous or of its own are captured and transported to peripheral lymphoid organs.
One of the mechanisms employed by the peripheral tolerance is achieved by the regulating cells of the immune system, called Tregs, they delete directly the auto-reactive cells. Several cytokines can influence the activity of Tregs cells, negatively or positively. The main sponsor of the activity of Tregs are IL-2 and TGF-b, whereas cytokines that promote Th17 response type (pro-inflammatory), decrease the activation and function of Tregs [39].
The decision of the T cell to activate or be tolerant is determined by the characteristics of the antigen and the regulation in the responses of the T cell mediated by the presence of different cytokines.
The beginning and the pathological consequences of the autoimmune diseases are often associated with the presence of alterations in the functional balance between pro-inflammatory cytokines (Th1 and Th17) and immune-regulators / modulators (Th2) [40].
Conclusions
Despite the efforts to recognize the target of autoimmunity in this pathology, it is still unknown. The question is whether these citrullinated residues present in excess could lead to a loss in tolerance, and if so, what process of the phenomenon of tolerance is affected?
If the autoimmune response is being generated to a secondary process caused cell death, we should ask ourselves if the recognition of these antigens is indeed a type of autoimmune response or an effort of the immune system to get rid of cells that are under stress situations .
Yet it is needed to characterize the inflammatory exacerbated response in these patients, in terms of priority, in other words, Is the presence of a pro-inflammatory response necessary for RA to be generated? o The presence of inflammation is secondary to the onset of the disease, and the same in terms of involvement of auto-antibodies, are they a triggering phenomenon? or consequence of the disease.
The globally comprehension about the origin, development and function of the different cell types involved in the generation of autoimmunity and their contribution to cause a loss of tolerance, will generate valuable knowledge for the development of new therapeutic options.
388
References
- WJ, K., Arthritis and allied conditions. 2001, Lippincot Williams and Wilkins: Philadelphia.
- Morales-Romero, J., La atención medica en reumatología en un hospital de segundo nivel de atención. Reumatol Clin, 2005. 1(2): p. 87-94.
- Sokka, T., et al., Functional disability in rheumatoid arthritis patients compared with a community population in Finland. Arthritis Rheum, 2003. 48(1): p. 59-63.
- Pearson, T.A., Where do we go from here? Am J Med, 2008. 121(10 Suppl 1): p. S32-4.
- Weyand, C.M. and J.J. Goronzy, Pathogenesis of rheumatoid arthritis. Med Clin North Am, 1997. 81(1): p. 29-55.
- Knedla, A., E. Neumann, and U. Muller-Ladner, Developments in the synovial biology field 2006. Arthritis Res Ther, 2007. 9(2): p. 209.
- Mattey, D.L., et al., Association of DRB1 shared epitope genotypes with early mortality in rheumatoid arthritis: results of eighteen years of follow-up from the early rheumatoid arthritis study. Arthritis Rheum, 2007. 56(5): p. 1408-16.
- van Gaalen, F.A., et al., Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum, 2004. 50(7): p. 2113-21.
- Zanelli, E., F.C. Breedveld, and R.R. de Vries, HLA class II association with rheumatoid arthritis: facts and interpretations. Hum Immunol, 2000. 61(12): p. 1254-61.
- Deighton, C.M., et al., The contribution of HLA to rheumatoid arthritis. Clin Genet, 1989. 36(3): p. 178-82.
- Takemura, S., et al., Lymphoid neogenesis in rheumatoid synovitis. J Immunol, 2001. 167(2): p. 1072-80.
- Weyand, C.M., et al., Lymphoid microstructures in rheumatoid synovitis. Curr Dir Autoimmun, 2001. 3: p. 168-87.
- Mauri, C. and M.R. Ehrenstein, Cells of the synovium in rheumatoid arthritis. B cells. Arthritis Res Ther, 2007. 9(2): p. 205.
- Firestein, G.S., Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol, 2005. 11(3 Suppl): p. S39-44.
- Nandakumar, K.S. and R. Holmdahl, Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther, 2006. 8(6): p. 223.
- van Boekel, M.A., et al., Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res, 2002. 4(2): p. 87-93.
- Zeng, X., et al., Diagnostic value of anti-cyclic citrullinated Peptide antibody in patients with rheumatoid arthritis. J Rheumatol, 2003. 30(7): p. 1451-5.
- Schellekens, G.A., et al., The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum, 2000. 43(1): p. 155-63.
- Herold, M., et al., Anti-CCP: history and its usefulness. Clin Dev Immunol, 2005. 12(2): p. 131-5.
- Vossenaar, E.R. and W.J. van Venrooij, Citrullinated proteins: sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res Ther, 2004. 6(3): p. 107-11.
- Schellekens, G.A., et al., Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest, 1998. 101(1): p. 273-81.
- Nielen, M.M., et al., Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum, 2004. 50(2): p. 380-6.
- Kastbom, A., et al., Anti-CCP antibody test predicts the disease course during 3 years in early rheumatoid arthritis (the Swedish TIRA project). Ann Rheum Dis, 2004. 63(9): p. 1085-9.
- van Gaalen, F.A., et al., Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum, 2004. 50(3): p. 709-15.
- Vossenaar, E.R., et al., The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum, 2004. 50(11): p. 3485-94.
- Makrygiannakis, D., et al., Citrullination is an inflammation-dependent process. Ann Rheum Dis, 2006. 65(9): p. 1219-22.
- Verpoort, K.N., et al., Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum, 2006. 54(12): p. 3799-808.
- Takizawa, Y., et al., Peptidylarginine deiminase 4 (PADI4) identified as a conformation-dependent autoantigen in rheumatoid arthritis. Scand J Rheumatol, 2005. 34(3): p. 212-5.
- Curis, E., et al., Almost all about citrulline in mammals. Amino Acids, 2005. 29(3): p. 177-205.
- Vossenaar, E.R., et al., PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays


