Key words
|
| |
| Griseofulvin, Nanocrystals, Nanocrystallization, Stabilizers, Bioavailability, Dissolution velocity |
| |
Introduction
|
| |
| Together with membrane permeability, the solubility/dissolution behavior of a drug is a key determinant to its oral bioavailability, the latest frequently being the rate-limiting step to absorption of drugs from the gastrointestinal tract. Since an increasing number of newly developed drug candidates present poor water-solubility, approaches to overcome this factor are of great importance in drug formulation (Hecq). At present about 40% of the drugs being in the development pipelines are poorly soluble, even up to 60% of compounds coming directly from synthesis are poorly soluble (Merisko- Liversidge). The solubility of a solute is the maximum quantity of solute that can dissolve in certain quantity of solvent or quantity of solution at a specified temperature. |
| |
| Aqueous solubility is one of the key determinants in development technologies, such as combinational chemistry and high throughput screening are based on the basic principles of medicinal chemistry, teaching that the most reliable method to increase in vitro potency is to add lipophilic moiety at appropriate positions of the lead structure. This has led to an increase in number of lipophilic and poorly soluble molecules being investigated for their therapeutic activity. Various formulation techniques are applied to compensate for their insolubility slow dissolution rate consequently poor therapeutic efficacy. These include formulation of the amorphous solid form, nanoparticles, microemulsions, solid dispersions; melt extrusion, salt formation and formation of water soluble complexes (Loftsson et al...2005). |
| |
| In the present study the area of interest are drugs belonging to class II of BCS classification. Class II consists of water-insoluble drugs which, when dissolved, are well absorbed from the gastrointestinal tract. The dissolution rate is usually the rate-limiting step in drug absorption. Commonly drugs in this class have variable absorptions due to the numerous formulation effects and in vivo variables that can affect the dissolution profile. Thus, a formulator by employing various formulation techniques tries to move the drugs from Class II to Class I without changing the intrinsic ability of the drug molecules to permeate biomembranes (Loftsson et al...2002). Drug delivery has become increasingly important mainly due to the awareness of difficulties associated with a variety of old and new drugs such as Clotrimazole, Nifedipine and Felodipine have motivated the development of drug delivery technologies to overcome the obstacles to their solubilization through either chemical or mechanical modification of the environment surrounding the drug molecules or physically altering the macromolecular characteristics of aggregates drug particles (Hu et al..2004). There are various other approaches to solubilize poorly soluble drugs, such as inclusion complexes with cyclodextrins, formation of mixed micelles and liposome formulation. However, these approaches have proved to be inefficient as evidenced by the low number of marketed products (Akkar et al...2004). Nowadays an alternative approach is being applied that is nanonisation. The drug powder is transferred to drug nanocrystals which are a new carrier free colloidal drug delivery system with particle size ranging from 100-1000 nm and is thought as a viable drug delivery strategy to develop the poorly soluble drugs, because of their simplicity in preparation and general applicability. The drug nanocrystal production techniques are classified as ‘bottom up’ methods and ‘top down’ methods (Busharb FM et al., 2006; Gao et al...2008). the common methods for preparing drug nanocrystals depend on advanced milling techniques that utilize excipient stabilization. Nanosuspensions produced by high pressure homogenization have recently been demonstrated to produce crystalline drug nanoparticles. Nanoparticle precipitation by the anti-solvent method is also a direct and simple procedure for the preparation of drug nanocrystals. However, it is usually difficult to control the particle size in the submicron region and the addition of surfactant as stabilizer is necessary to avoid the formation of microparticles. Cyclodextrins are being now a days being more widely used for the non-inclusion based aspect of interaction between them and drug molecule, including formulation of molecular aggregates and surfactants-like effects.(Makhlof et al..2008). Sodium lauryl sulfate is also used as stabilizing system in the formulation of nanosuspensions (Eerdenburgh BV et al...2008). The aim of the present work is to enhance solubility and dissolution rate of griseofulvin a class II drug by converting it into nanocrystal by emulsion solvent diffusion method/ solvent displacement/ nanoprecipitation in the presence of β-cyclodextrin and Sodium lauryl sulfate as nanocrystal stabilizer. |
| |
|
Material
|
| |
| Griseofulvin (Pharma Grade) (Cosme pharma, Goa), PVA (Pharma Grade) (West Coast Laboratory. Mumbai), Ethanol (Merck. Mumbai), Sodium lauryl sulphite (S.D Fine chem. Mumbai), β-Cyclodextrin (Apex Health Care Ltd, Gujarat). |
| |
Methodology
|
| |
|
Preparation of griseofulvin nanocrystals
|
| |
| The nanocrystals were prepared by emulsion solvent diffusion method. This method involves the use of an organic phase, which is completely soluble in the external aqueous phase. The organic solvent containing drug diffuses instantaneously into the external aqueous phase inducing immediate precipitation of drug as nanocrystals because of complete miscibility of both phases. This method is particularly applicable for highly hydrophobic drugs which can be precipitated as nanocrystals in aqueous phase (Vyas SP et al., 2008). Β-Cyclodextrin and sodium lauryl sulphate (SLS) were used as suspension stabilizers to prevent aggregation of nanocrystals. |
| |
| Typically the polar organic drug solution was prepared by dissolving the drug in polar organic solvents. Different concentrations of drug solution were prepared. The polar organic drug solution was drained into 0.1% aqueous solution of β-Cyclodextrin and sodium lauryl sulphate (SLS) at a rate of 2ml per min with continuous stirring at 400 rpm with propeller mixer. The formed dispersion was immediately centrifuged and the residue was redispersed in distilled water with sonication. The process was repeated twice and final dispersion was subjected to lyophillization. The list of formulations and the concentrations along with stabilizer concentration is shown in table 1. |
| |
|
Particle size analysis
|
| |
| Size and size distribution of the crystals in dried form was determined following redispersion in water containing 0.1% polyvinyl alcohol (PVA-403) by dynamic light scattering through particle size analyzer Nanotrac 150 (Japan) with a wet sampling system and the diameters reported were calculated using mean particle size distribution (Makholf A et al., 2008). |
| |
|
Determination of griseofulvin content
|
| |
| The drug content of freeze dried samples was checked by UV-spectrophotometer to confirm the purity of the prepared samples. For quantitative determination of Griseofulvin content in formulations aqueous dispersions of formulations (25mg/10ml distilled water) were passed through 0.8 μm filter. The filtrates containing fine particles smaller than 0.8 μm were dissolved in 4% sodium lauryl sulfate solution and the concentration of griseofulvin was determined spectrophotometrically at a wavelength of 291 nm (UV-1201 Shimadzu Corporation, Japan). The amount of drug in filtrate relative to the total amount of drug in the dispersion was calculated and expressed as nanocrystal yield (Makholf A et al., 2008). |
| |
|
Scanning electron microscopy
|
| |
| The surface morphology of the commercial griseofulvin powder and the freeze-dried formulation samples was examined by SEM (JEOL-JSM-T330A Nihon Denshi, Japan). Before examinations the samples were mounted on top of double sided sticky carbon tape on metal discs and coated with 80 nm gold/palladium in Balzers 120B sputtering device (Waard H et al., 2008). |
| |
|
Powder X-ray diffraction (PXRD)
|
| |
| The PXRD was carried out using Philips Analytical XRD B.V. at the scanning rate of 40/min 2θ range of 10-70°. |
| |
|
Differential scanning calorimetry
|
| |
| DSC (DSC-60, Shimadzu, Tokyo, Japan), equipped with a liquid nitrogen cooling system was used to measure the thermal behavior of the commercial griseofulvin powder and the freeze dried samples. In DSC analysis, 2-5 mg of sample was put in aluminum pan and examined at a scanning rate of 100 C/min from 25 to 300°C. |
| |
|
Solubility
|
| |
| Saturation solubility measurements were assayed through ultraviolet absorbance determination at 291 nm using an UV spectrophotometer. An excess amount of griseofulvin powder and formulations were added to 150 ml of 4% SLS solution the mixture were stirred in mechanical shaker for 24 hours at a temperature of 37+ .05°C using GLF 1086 shaker (Labortechnik, Burgwedel, Germany). Visual inspection was carefully made to ensure there was excess sample in solid state indicating that saturation had been reached. The mixtures were filtered using 0.2μm filter and filtrates were diluted suitable to determine the solubility of griseofulvin from each formulation. |
| |
|
Dissolution test
|
| |
| A dissolution test (USP XXIII Electro Lab TDT-80) for commercially available griseofulvin and formulations was carried out by filling them in hard gelatin capsules (Zydus Cadilla, Goa, India). The prepared samples and the drug powder were filled in capsules (125mg) and subjected to dissolution studies with 900 ml 4% SLS solution as dissolution medium preheated and maintained at 37 + 0.5°C. The baskets were rotated at a speed of 75 rpm/min. 10ml samples were withdrawn at specified time intervals, filtered through 0.2μm filter, and the concentration of griseofulvin was determined by UVspectrophotometry. |
| |
|
Stability studies
|
| |
| All the formulations were subjected to stability study as per ICH guidelines the formulations were divided into two parts and stored at 30o± 2°C and 65% ± 5% RH and 40°± 2°C and 70% ± 5% RH. The drug release and the drug content were estimated after specified intervals of time. (Given in table 3) |
| |
RESULTS
|
| |
| When the drug solution of ethanol and acetone was added to an aqueous phase in the presence of cyclodextrin and SLS by emulsion solvent diffusion method, a homogenous dispersion was obtained. Sticking of griseofulvin particles to the propeller was observed in the absence of stabilizers. The dispersion was centrifuged in order to yield an easily redispersible pellet of particles. The particles were washed thoroughly to remove traces of organic solvent by repeated redispersion and centrifugation. Finally the dispersion in water was freeze dried. A total of eight formulations were prepared with two different solvents and two different stabilizers. The drug solution concentration was also considered during the formulation. |
| |
| Dynamic light scattering method was used to determine the particle size of freeze dried samples. Table 1 shows the average particle size values for griseofulvin nanocrystals prepared by emulsion solvent diffusion in different solvents and different stabilizers. Submicron sized particles with average diameters in the range of 600-900 nm were successfully produced. |
| |
| The particle surface morphology of freeze dried samples of formulations and griseofulvin powder was characterized by the scanning electron microscopy (SEM). The pure griseofulvin powder as acquired was in the form of needle shaped crystals. Over the whole set of formulations, griseofulvin was precipitated as crystalline product, with morphology of needle or bipyramide as shown in fig1. |
| |
| The crystal property of griseofulvin powder and freeze dried formulations was evaluated by powder X-ray diffraction analysis fig 2. |
| |
| The diffractograms confirms that the method for preparing nanocrystals does not interfere with griseofulvin crystalline state as diffraction patterns are conserved for the formulated nanocrystal formulations. |
| |
DISCUSSION
|
| |
| In this study an attempt was made to process griseofulvin powder in the form of drug nanocrystals, they were prepared by emulsion solvent diffusion method. Two solvents acetone and ethanol were used in the formulation and two different stabilizers β-cyclodextrin and SLS (0.1%) were used to keep the nanocrystals segregated from each other and avoid aggregation. Griseofulvin was chosen as model drug because it possesses poor aqueous solubility but good permeability because of which it shows very unpredictable bioavailability (Tripathi KD 2008). With this method crystalline dispersions of nanosize range were successfully prepared and freeze dried in a reproducible manner. |
| |
| The particle size of the freeze dried samples was determined using dynamic light scattering method after dispersion in distilled water by sonication for 5min. When only distilled water was used to disperse the nanocrystals, larger particles in range of 3-5 μm could be observed. However, samples dispersed in 0.1% polyvinyl alcohol (PVA-403) exhibited a monodispersed particle size distribution in the nanosized range. This behavior exhibited that the freeze dried formulations are aggregates of submicron particles. Griseofulvin content of the freeze dried formulations was determined quantitatively by UV-spectrophotometry and the results confirmed the purity of the prepared samples. The amount of drug recovered after filtration through 0.8 μm filter was determined and expressed as % of the total griseofulvin present in formulation. The formulations prepared using β-cyclodextrin showed better drug content than with SLS. This could be due to the ability of β-cyclodextrin to form a network through intermolecular interaction that could protect and stabilize the produced nanocrystal. It was reported that β-cyclodextrin can self-associate in aqueous solutions to form nano-scale aggregates that have a minimum hydrodynamic radius. SLS is also used as dispersion stabilizer in this work which prevents agglomeration of precipitated nanocrystals in the formulation by increasing the activation energy of the process. It reduces the surface tension existing between the drug particle and the solvent by providing wettability to the particles. As SLS is an ionic surfactant the possible mechanism of providing barrier is through electrostatic charge on the surface of the particles. |
| |
| Crystals have usually well-defined faces. Fractures and breaks can be observed in the SEM images which could be attributed to the stirring turbine especially when long needles were recovered. |
| |
| The change of morphology from starting material raised the question of possible polymorphism. Because polymorphs differ in the type of lattice, or in spacing of the lattice points, they can exhibit different crystalline shapes. Crystals of different habit have identical physical properties, whereas polymorphs on the other hand, exhibit different physical properties, as density, hardness, reactivity, thermal properties, optical behavior, etc. Needles and bipyramids exhibited similar X-rays diffractogram patterns close to the starting material. The PXRD patterns of pure Griseofulvin and formulations F1A and F2A are presented in fig 3. The PXRD patterns of pure Griseofulvin showed numerous sharp peaks, which are the characteristic of a crystalline compound. Drug crystallinity peaks were also detectable in formulations as shown in fig 5.4 to 5.6. Polymorphism is the capability of a substance to crystallize into two or more different forms. |
| |
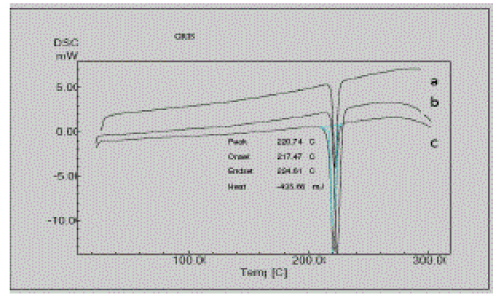
| Any polymorphic changes in the drug may change its melting point, bioavailability, and release kinetics. The polymorphic change in the drug griseofulvin was also studied using differential scanning calorimetry by testing the melting characteristics of the drug. Figure 4 gives a comparison of the thermograms of pure griseofulvin and formulations F1A and F1B. |
| |
| Griseofulvin showed a large and sharp characteristic endothermic peak at 220°C due to its phase transition. The onset and endset of phase transition of griseofulvin were observed at 217.61°C and 224.68°C. DSC thermograms of the formulations F1A and F2A showed characteristic endothermic peaks at 222.10°C and 223.73°C respectively. The DSC thermograms of formulations F1A and F2A showed characteristic endothermic peaks corresponding to those of the pure drug and there is no appearance of one or more new peak or disappearance of one or more peak corresponding to those of the pure drug. This indicates that crystalline nature remains with slight change in crystallinity due to change in solvent systems evident form minor shifts form melting point. Besides this, no additional peaks to demonstrate the significant changes in the melting characteristics of griseofulvin in the formulations were found indicating no polymorphic changes in griseofulvin in both formulations. The peaks were found to be nearly identical, with a calculated TH of pure drug, F1A and F2A were around -436.72 mJ, - .224.73 mJ, and -506.10 mJ respectively. |
| |
| It is generally acknowledged that drug nanocrystals offer a variety of physicochemical advantages over the large sized drugs, including the possibility for increased solubility, bioavailability and physical dispersion stability. Buckton and Beezer assumed that the enhancement of solubility is valid only for sparingly soluble particles of less than 1 μm in size (Buckton et al., 1992). As reported before, the particle size average of re-dispersed griseofulvin nanocrystals was below 1μm. Therefore, theoretically griseofulvin drug nanocrystals can overcome the bioavailability problem associated with low aqueous solubility. The solubility of griseofulvin nanocrystal formulations in 4% SLS medium increased by ten folds when compared with the solubility of griseofulvin. Similarly the solubility in water also increased by 6 folds as shown in table 2. According to the crystalline state investigation, the solubility enhancement of the griseofulvin nanocrystals is not due to the presence of the griseofulvin in a morphous form but due to the particle size reduction to the submicron range. This result is in agreement with the Kelvin–Gibbs equation and Ostwald–Freundlich equation (Buckton et al., 1992. Wenju W et al., 1998). All the formulations of griseofulvin were subjected to in vitro release of pure drug griseofulvin. |
| |
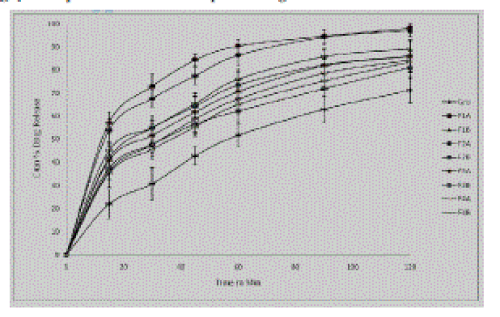
| These in vitro release studies were carried out using 4% SLS solution as dissolution medium. From the results of in vitro study it was observed that the batch F1A and F2A gave highest % cumulative drug release which might be due to the addition of low concentration of drug in solvent and secondly due to use of acetone as solvent than in formulations F3A to F4B where ethanol was used as solvent. Batches F1A to F4B gave the %drug release of 98.17, 89.21, 97.14, 86.09, 85.99, 81.14, 84.42, and 83.47 respectively. From this study it was found out that, as the concentration of drug in solution decreased drug release from formulations increased. And also the type of solvent used also affected the drug release form the formulation. These findings were supported by study that acetone generally yielded less particle size and increased release rate (Prasad et al 2007). It was found that cumulative percent drug release of all formulations was increased due to particle size reduction as compared to pure drug after two hours. Further the use of acetone as solvent in formulation F1A and F2B showed increased release rate of griseofulvin when compared to other formulations and pure drug. |
| |
| The initial burst release of griseofulvin can be due to small particle size and also due to use of 4% SLS solution as dissolution medium. The presence of SLS in dissolution medium lowers surface tension around the particle and helps them to dissolve rapidly. Some drugs are well absorbed in fed state and griseofulvin is one such drug. Use of 4% SLS solution mimics the fed state of stomach (Balakrishanan A et al 2004). The enhanced dissolution property of nanocrystals can be described by Noyes-Whitney equation. |
| |
| Dc/dt= D — A/h(cs − cx) |
| |
| Where, D is the diffusion coefficient, A the surface area, cs is the saturation solubility, cx the bulk concentration, and h is the so-called “diffusional distance” over which the concentration gradient occurs. It is obvious that an increase in the surface area consequently increases the dissolution velocity. |
| |
| In addition, drug nanoparticles are characterized by an increase in saturation solubility cs. According to Noyes–Whitney, the increase in cs—in addition to the enlarged surface area—further increases the dissolution velocity. The dissolution velocity is reversely proportional to the diffusional distance h, which means that reducing h leads to a further increase in dissolution velocity. According to the Prandtl equation (Müller et al., 2004), the diffusional distance h decreases for very small particles. The simultaneous increase in saturation solubility cs and decrease in h leads to an increased concentration gradient (cs−cx)/h, thus enhancing the dissolution velocity in addition to the surface effect Dissolution studies of the lyophilized griseofulvin nanocrystals clearly showed this advantageous phenomenon. Dissolution velocities of the dried griseofulvin nanocrystals were distinctly superior compared to the raw material. |
| |
CONCLUSION
|
| |
| In this study it was found that nanocrystallization is a good approach in enhancing dissolution property of poorly soluble drugs by emulsion solvent diffusion method. Acetone and ethanol were used in this study as solvents for drug out of which formulations made of acetone were of smaller size compared to ethanol. Formulations made by using acetone as solvent also possessed better dissolution velocity as compared to formulations prepared using ethanol as solvent. The toxic effects of these formulations finding their way into final formulations was restricted by repeated washing of the pellets after centrifugation of the dispersion for several times. Two stabilizers namely β-cyclodextrin and SLS were used to stabilize the dispersions. |
| |
| Although there were no significant differences in terms of particle size in the formulations, still the effect of concentration of these stabilizers on particle size, its distribution and dissolution velocity needs to be studied. Thus nanocrystal drug delivery system can adopted to increase the solubility and dissolution rate of poorly soluble drugs to enhance their bioavailability. |
| |
Tables at a glance
|
|
|
| |
Figures at a glance
|
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
|
| |


