Keywords
Purα; U87MG cell; γH2AX; stable cell lines; DNA damage
Introduction
Human transcriptional activator alpha (Purα) is a ubiquitous nucleic acid-binding protein that is originally purified from the mouse brain based on its ability to bind to a DNA sequence derived from the promoter of the mouse myelin basic protein gene [1,2]. Human Purα is characterized by the ability to bind to a DNA sequence presented upstream of the human c-myc gene, its DNA is cloned from HeLa cells and sequenced already [3,4]. The sequence of mouse Purα [5] and human Purα [4] are nearly identical with only 2 out of 322 amino acid residues differed. The DNA binding domain of Purα is strongly conserved throughout evolution. Purα is a member of Pur family of proteins along with Purβ and Purγ, for which two isoforms exist; that rise from the usage of alternative polyadenylation sites[6]. Purα is expressed virtually in every metazoan tissue [7] and it is a multifunctional protein that can bind to both DNA and RNA and functions in the initiation of transcription, DNA replication, DNA repair and RNA transport [7-9].
Purα knockout (Purα-/-) mice were genetically engineered [10]. The mice were normal at birth, but they developed neurological problems with severe tremor and spontaneous seizures at the 15th postnatal day; they died in the 4th week after birth. There were lower numbers of neuron in regions of the hippocampus and cerebellum in Purα-/- mice versus those of age-matched wildtype (Purα+/+) littermates, suggesting that Purα plays a critical role of neurogenesis [11]. The straitened circumstances for the investigation of the functions of Purα in nervous system lie in the difficulties to obtain the adult mice with Purα gene knocked out since the Purα knockout mice could not live to their maturated ages. The cell lines originated from nervous system is relatively difficult for the transfection with the normal approaches. The lower efficiency of transfection cannot match the requirement of most experiments of gene manipulations. To deliver exogenous DNA into cells with electroporation is a common method for DNA manipulation but the problem consisted in the cell death and adjustment of the optimal condition for the experiments, so it is not easy to keep the consistency of each experimental conditions. The recent developed approaches of infection of cells with lentivirus can get better transfection efficiency and it is helpful for the experiments with gene manipulation, but the construction of lentivirus and packaging of the virus usually expends a lot of time and energy and the variances of virion titers also affects the consistency of the experiment. In order to overcome the disadvantages existed in the above mentioned approaches of gene transfection and to obtain the uniform cell lines for the research, we reconstructed the lentivirus plasmid pLKO.1 and established the constructs which can overexpress fluorescent protein, pLKO.1-puro-GFP as well as that fused with Purα, pLKO.1- puro-EGFP-Pura. The reconstructed plasmids were packaged and the obtained virions were used to infect human glioblastoma cell line U87MG and the cells were cultured under the suppression of puromycin to select the stable cell lines in which the exogenous genes of EGFP and EGFP-Purα were successfully integrated into the cellular genomic DNA. The established cell lines provide a useful tools for our ongoing and incoming experiments.
A numerous endogenous and exogenous agents, which include exogenous endotoxins such as ionizing radiation (IR), ultraviolet (UV) light, chemotherapeutic agents, as well as endogenous associated with the metabolites of oxidation, stalled DNA replication and V (D) J recombination, can cause DNA damages such as single-strand break (SSB) or double strand break (DSB) which would affect the integrity of genomic materials inside the cells. A remarkable symbol of DNA damage is the phosphorylation of H2AX to form the phosphorylated H2AX, usually named as γH2AX, in this way H2AX is considered as the symbolic protein for DNA damage [12]. The established cell lines were verified by a series of experiments and the model of DNA damage was established by application of hydroxyurea (HU) to the established cell lines and the function of Purα in DNA repair has been investigated.
We present here the results of detailed molecular and functional studies which suggest the role of Purα in DNA repair, an important function on Purα in maintenance of the integrity and stability of the genomic DNA in cells.
Materials and Methods
Materials
Lentivirus plasmid pLKO.1-puro was purchased from Addgene, fluorescent expression vector pEGFP-C1 vector was purchased from Clontech, eukaryotic expression vector pCDNA3.0-Purα is kept in our lab, cell lines HEK 293FT and U87MG are kept in our lab, CCK-8 kit was purchased from Bestbio; Puromycin was purchased from InvivoGen, Hudroxyurea was purchased from SIGMA; Purα, Grb2 and GAPDH antibodies were purchased from Abcam; fluorescent antibody IRDye®800CW was purchased from LI-COR
Construct pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα lentivirus plasmids
Construction of lentivirus plasmids were illustrated in Figure 1. In short, to amplify full length Purα gene with PCR using pCDNA3.0- Purα as template, BglII and Eco RI endonuclease sites were added to the both sides of the Purα DNA. Then the full length Purα DNA with the designed endonuclease sites was sub-cloned into the multiple cloning site with same endonuclease site of fluorescent expression vector pEGFP C-1 and obtained the intermediate cloning vector pEGFP-C1-Purα, then cutting with AgeI and EcoRI to get the EGFP-Purα fragment and sub-cloned it into the multiple cloning site of lentivirus plasmid pLKO.1-puro to get the pLKO.1-puro-EGFP-Purα plasmid. In the same way to cut the EGFP fragment from pEGFP-C1 and insert it into pLKO.1-puro plasmin to get the pLKO.1-EGFP plasmid which is used as control of empty vector in the afterward experiment.
Cell culture and package of lentivirus
HEK293FT and U87MG cells were maintained with DMEM medium supplemented with 10% FBS, 100U/ml penicillin and 100U/ml streptomycin and kept in the incubator at 37°supplemented with 5%CO2. Cells were cultured in 6-well plates when cell counting assay was performed and HU was added to the culture medium at the final concentration of 2mM and continued to culture for 8 hours to harvest the cells. The cell total RNA and proteins were extracted for the further experiments. Virus packaging was performed according to the manufacturer’s instruction. In short, the constructed lentivirus plasmids and packaging plasmids pCMV-dR8.2 dvpr and pCMV-VSV-G were co-transfected into HEK 293 FT cells according to the following ratio:
pLKO.1-puro-EGFP (pLKO.1-puro-EGFP-Purα)1000ng
pCMV-dR8.2 dvpr 900ng
pCMV-VSV-G 100ng
To change the normal medium into the medium without antibiotics before the transfection, using Lipofectamine® 2000 (Invitrogen) to do the transfection according to the manufacturer’s instruction. 8 hours after the transfection to change the medium into the normal fresh medium and continued to culture the cell for 24 and 48 hours to collect the medium respectively for virus purification. To centrifuge the medium at 1500rpm for 5 min to remove the cell debris, the supernatant of the collected medium was centrifuged at 15000rpm, 4, overnight to precipitate the virion. The virion sediments was resolved in DMEM and kept in -80, for the future use. The titter of the virus was measured with flow cytometry. 90μl of HEK 293FT cells (1X104 cells) were seeded in the 96-well cell culture plate and the virion solution was sequentially diluted with 1:3 dilution ratio and add 10 μl of virion to each well. Cells were cultured for 48 hours and applied to flow cytometry for titer assay.
Selection of the concentration of antibiotics
U87MG cells ware split and cultured in 24-well cell culture plates at the density of 1.0X105/well. When the cell confluence reached to 80%-90%, the different concentrations of puromycin were added to the culture medium and make the final concentration to be 0, 1, 2.5, 5, 7.5 and 10μg/ml respectively. Cells were cultured under the above condition for 4-7 days and to change the medium every 2 days. The concentration of puromycin which can lead to all cell die within 3 to 4 days was chosen as the selected concentration.
Establishment of the stable cell lines
The constructed lentivirus plasmids, pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα were used to be transfected into U87MG cells, to change medium with 5.0μg/ml puromycin in 48 hours after the transfection to select the cells of which the resistance of anti-puromycin were obtained. The fluorescent protein expressed in the survived cells were checked under fluorescent microscope. The survived cells were trypsinized and performed the further culture with limited dilution method and the single colony was selected for the further culture to establish the stable cell lines of pLKO.1-EGFP and pLKO.1-EGFP-Purα.
Real-time PCR to check the expression of Purα in established stable cell lines.
To extract total RNA from the established cells with Trizol and then to do the reverse transcription with RevertAid First Strand cDNA Synthesis Kit (Thermo) according to the manufacturer’s instruction. After the reverse transcription, using the transcripts as templates to synthesize the first strand of cDNA. The following primers were used to check the expression of Purα in the established cell lines. RT-PCR for Purα: Primer-F: 5’- CGT GTT TAT GCG AGT GAG-3’, Primer-R: 5’- CCT CTG CTT CTC TTG AATC-3’; RT-PCR for GAPDH:
Primer-F: 5’- GAG TCA ACG GAT TTG GTCGT-3’ Primer-R: 5’-GAC AAG CTT CCC GTT CTCAG-3
CCK-8 cell proliferation and cytotoxicity test
CCK-8 cell proliferation and cytotoxicity test is a susceptibility testing and the cell proliferation rate were determined by a colorimetric cell viability assay based on reduction of a tetrazolium salt {2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8)} via 2-methyl-1, 4-napthoquinone as an electron mediator. WST-8 can be deoxidized by dehydrogenase in the mitochondria to form aurantius- colored formazan. The darkness of the color is proportional to the cell numbers in the same cells. The WST-8 colorimetric assay is a useful method for rapid determination of cell proliferation and cytotoxicity. To compare with MTT method, it is characterized as more sensitive and simple. Cells were treated as single cell suspension and added 100ul of suspended cells into 96-well cell culture plate at the density of 1×104/well. The cells were maintained at incubator in 37, supplemented with 5%CO2 overnight. The 10μl of different concentrations of HU were added to the cells and made the final concentration to be 0, 0.25, 0.5, 1, 1.5, 2.0, 3.0mM respectively and continued to culture for 18 hours, and then add 10μl CCK-8 reagent and continued to culture the cells for another 1 hour. The blank group was set as the wells adding medium and CCK-8 reagent without cells, control group was set as cells with medium but without CCK-8 reagent. Each group of cell were set as quintuplicate. The OD450 of each well was measured in spectrophotometer.
Pulse field gel analysis
Chromosomal DNA damage in cells was determined by asymmetric field inversion gel electrophoresis (AFIGE), following procedures previously described [13,14]. Cells were plated in 6-well plates at 1 x 105 cells per well and treated with HU. The treated cells were collected, mixed in 0.5% agarose (InCert agarose FMC) and made in 3x5 mm cylinder plug containing 1x105 cells per plug. Cells in the agarose plugs were lysed in lysis buffer (10 mM Tris?HCl, pH 8.0, 50 mM NaCl, 50 mM EDTA, 2% N-laurylsarcosyl, 0.1 mg/ml proteinase E (Sigma) at 50?C for 16?18 hours, washed with 10 mM Tris?HCl, pH 8.0, 0.1 mM EDTA at 37?C for 1 hour, and digested with RNase buffer (10 mM Tris?HCl, pH 7.5, 0.1 mM EDTA, and 0.1 mg/ml RNase A. The treated plugs were loaded onto 0.5% agarose gel in 0.5X TBE buffer containing 45 mM Tris (pH 8.3), 45 mM Boric Acid, 1 mM EDTA containing 0.5 mg/ml ethidium bromide. Asymmetric field inversion gel electrophoresis was carried out for 40 hours with cycles of 1.25 V/cm for 900 seconds in the migration direction and 5 V/cm for 75 seconds in the reverse direction. The gel was imaged by photographing the ethidium bromide under UV light. The gel was analyzed using Image J software (Rasband, W.S., ImageJ, US National Institutes of Health, Bethesda, Maryland, USA, https://rsb.info.nih.gov/ ij/, 1997–2006). The fraction of DNA released (FDR) from the well into the lane is a measure of DSBs present. FDR measured in untreated samples was subtracted from FDR measured in samples treated with HU.
Western blotting
The established cell lines pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα were treated with 2mM HU for 18 hours and the cell proteins were extracted. The protein concentration was quantitated with Braford method. 60μg of total protein was applied to SDS-polyacrylamide gel electrophoresis for western blotting assay. The primary antibody anti-Purα used in the experiment was diluted in the ratio of 1: 1000, anti-GAPDH antibody was diluted as 1: 1000, Anti-Grb2 antibody was diluted as 1:1000, the ratio of fluorescent labeled second antibody was diluted at 1: 8000.
Data analysis
All the data were analyzed with SPSS20.0 statistical software and all the data were expressed as X±SD. The differences between each group were analyzed in one-way ANOVA. p<0.05 was set as significantly statistical differences.
Results
Successfully constructed EGFP-fused Purαlentivirus plasmid pLKO.1-puro-EGFP-Purα and pLKO.1-puro- EGFP
Lentivirus plasmids pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα were constructed successfully and identified by endonuclease cutting and proved to be consistent with the design and sequencing results demonstrated that the open reading frame is correct for the fusion protein. (Figure 1)
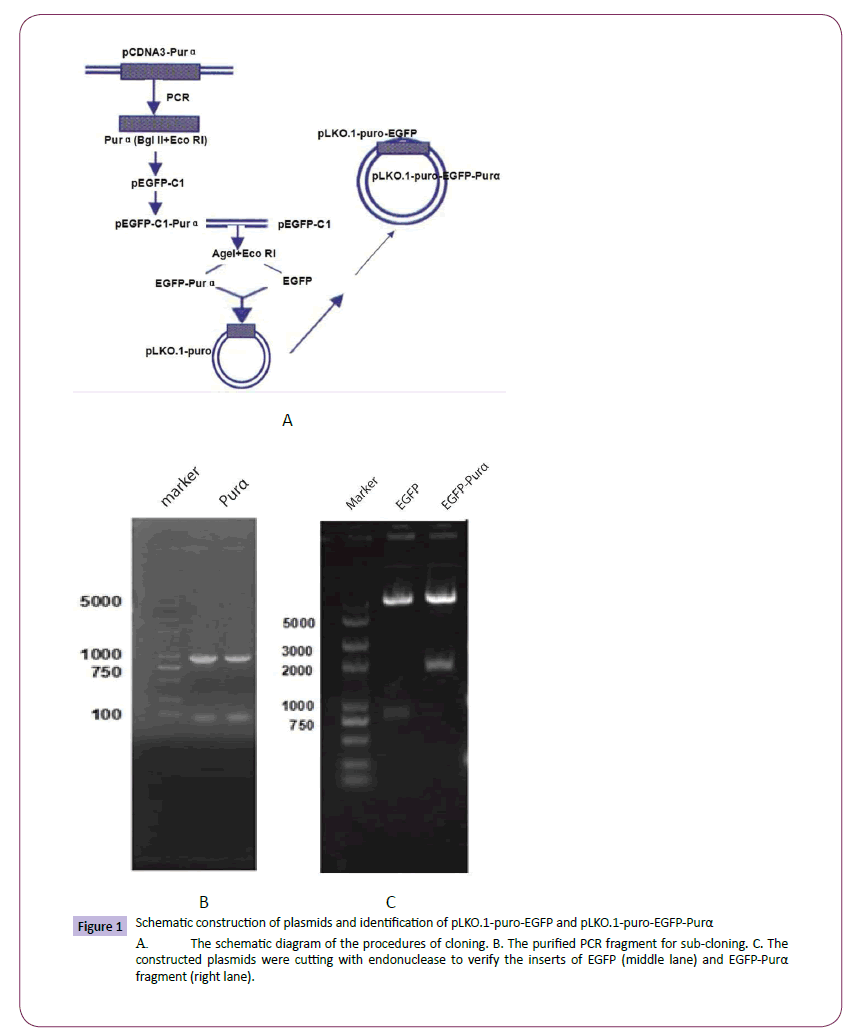
Figure 1: Schematic construction of plasmids and identification of pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα
A. The schematic diagram of the procedures of cloning. B. The purified PCR fragment for sub-cloning. C. The constructed plasmids were cutting with endonuclease to verify the inserts of EGFP (middle lane) and EGFP-Purα fragment (right lane).
Packaging virion and titers measurement
The titters of virion were detected with cytometry as described in the Materials and Methods above. The results demonstrated that titers of PLKO.1-EGFP-Purα was 6.26×107 and that of pLKO.1- EGFP was 5.89×107, the titers of the virion are enough to meet the experimental requirements. Figure 2
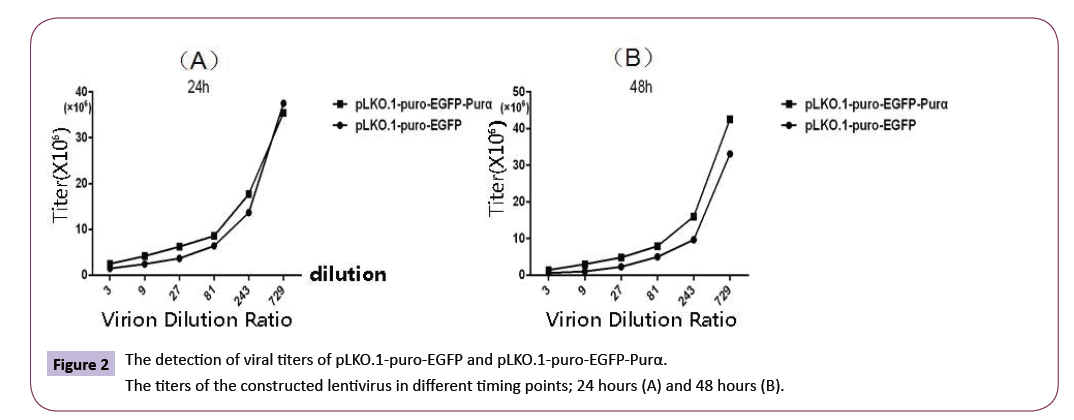
Figure 2: The detection of viral titers of pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα.
The titers of the constructed lentivirus in different timing points; 24 hours (A) and 48 hours (B).
Selection of the suitable concentration of antibiotics
The U87MG cells were treated with different concentrations of puromycin for 4 days and the status of the cells were checked under the microscope. The results demonstrated that when cells were treated with 2.5μg/ml puromycin, partial cells were dead, but when the puromycin concentrations were at 5.0, 7.5, 10μg/ ml, all the cells died. So we choose puromycin concentration of 5.0μg/ml as our selected concentration for the colony selection in the establishment of the stable cell lines (Figure 3).
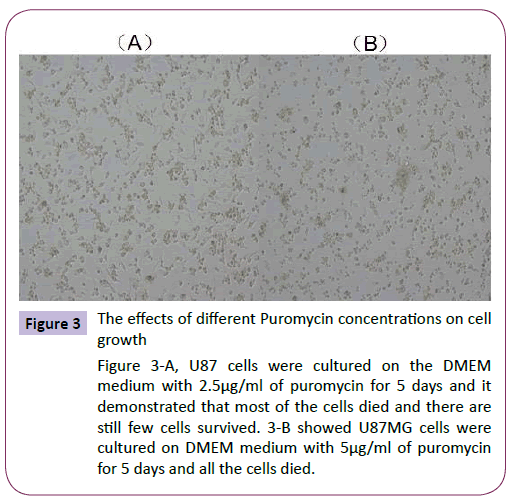
Figure 3: The effects of different Puromycin concentrations on cell growth
Figure 3-A, U87 cells were cultured on the DMEM medium with 2.5μg/ml of puromycin for 5 days and it demonstrated that most of the cells died and there are still few cells survived. 3-B showed U87MG cells were cultured on DMEM medium with 5μg/ml of puromycin for 5 days and all the cells died.
Establishment and identification of stable cell lines of pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα
The established cell lines were checked under the microscope to observe the fluorescent protein expression (Figure 4). The results demonstrated that Purα protein fused with EGFP appeared mainly in the cytoplasm. At the meantime the cell total RNA and proteins were extracted for real time PCR and western blotting assay to analyze the expression of Purα in the established cell lines. The results demonstrated that the expression of Purα in pLKO.1-puro-EGFP-Purα cells is much more than that in pLKO.1- EGFP cell lines (Figure 5). This results confirmed that the cell lines we established are successful and it can over-express Purα protein both in transcriptional and translational levels.

Figure 4: Identification of pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα cell lines under fluorescent microscope
4-A showed the pLKO.1-puro-EGFP cell lines under the fluorescent microscope and all the cells can express GFP protein. The GFP protein distributed both in nucleus and cytoplasm. 4-B showed the pLKO.1-puro-EGFP-Purα cell lines and all the cell express fluorescent proteins, but the EGFP-fusion-Purα proteins mainly appeared in the cytoplasm.
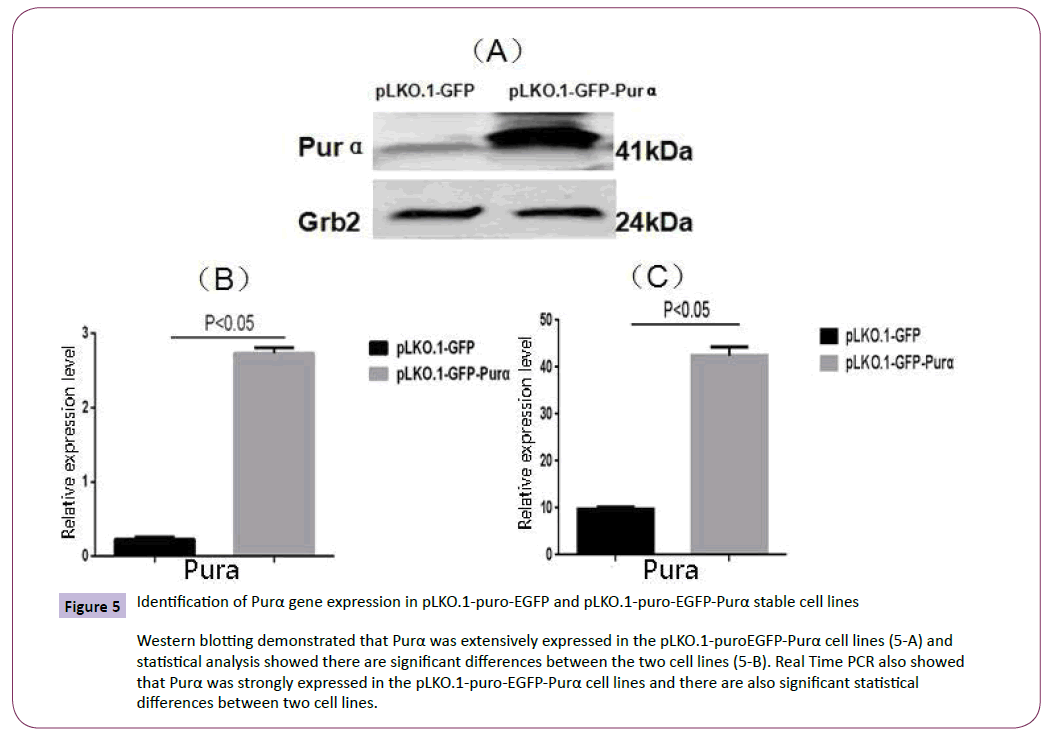
Figure 5: Identification of Purα gene expression in pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα stable cell lines
Western blotting demonstrated that Purα was extensively expressed in the pLKO.1-puroEGFP-Purα cell lines (5-A) and statistical analysis showed there are significant differences between the two cell lines (5-B). Real Time PCR also showed that Purα was strongly expressed in the pLKO.1-puro-EGFP-Purα cell lines and there are also significant statistical differences between two cell lines.
The effects of over-expression of Purα on cell proliferation in established cell lines
The established cell lines pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα were treated with different concentration of HU and the cell proliferation was analyzed with cell counting kit (CCK-8). The results demonstrated that along with the increment of HU concentration, the cell growth rate became slower, compared with the control cell line pLKO.1-EGFP , the Purα over-expression cell line,pLKO.1-puro-EGFP-Purα exhibited higher growth rate than that of the control group (Figure 6). This indicates that overexpression of Purα can alleviate the cytotoxicity of HU on cells and protect the cell from the DNA damage caused by HU.
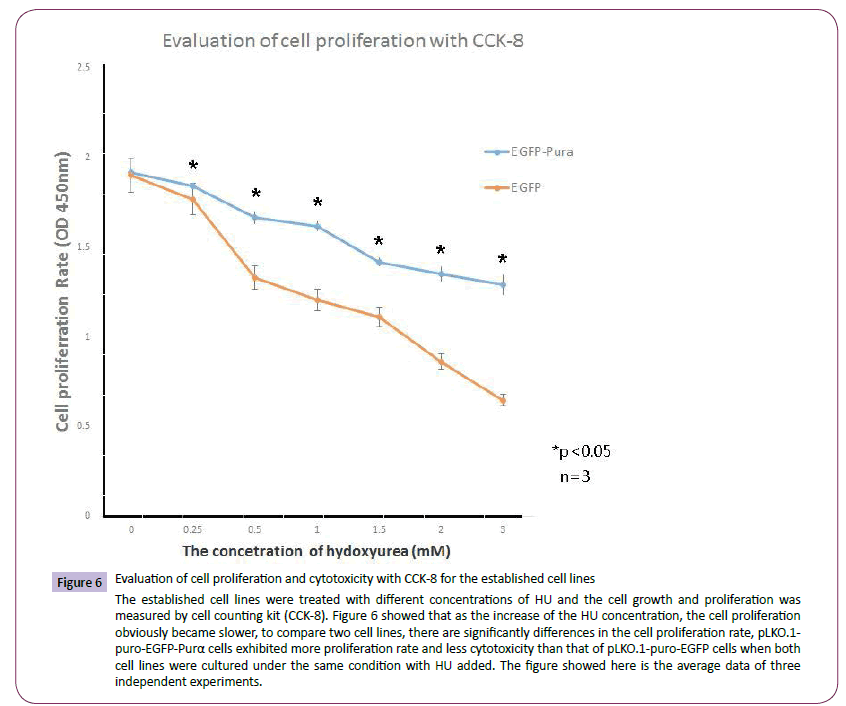
Figure 6: Evaluation of cell proliferation and cytotoxicity with CCK-8 for the established cell lines
The established cell lines were treated with different concentrations of HU and the cell growth and proliferation was measured by cell counting kit (CCK-8). Figure 6 showed that as the increase of the HU concentration, the cell proliferation obviously became slower, to compare two cell lines, there are significantly differences in the cell proliferation rate, pLKO.1- puro-EGFP-Purα cells exhibited more proliferation rate and less cytotoxicity than that of pLKO.1-puro-EGFP cells when both cell lines were cultured under the same condition with HU added. The figure showed here is the average data of three independent experiments.
The protective effects of Purα on cell DNA damage
The established cell lines pLKO.1-puro-EGFP and pLKO.1-puro- EGFP-Purα were treated with 2mM HU for 18 hours and then the cell total protein was extracted for western blotting assay of DNA damage symbol protein γH2AX and also for pulse field inversion gel electrophoresis assay to check the . The results demonstrated that in control group, the cells exhibited much more released DNA fragment than that of Purα over-expression group (Figure 7). The results of western blotting assay for γH2AX also demonstrated that compared with untreated group, the content of γH2AX is much higher in treated group, but in treated group, the amount of γH2AX is less in Purα overexpression group than that in the control group (Figure 8), there are significant statistical differences (P<0.05). These results demonstrated that Purα protein can alleviated the DNA damage caused by HU.
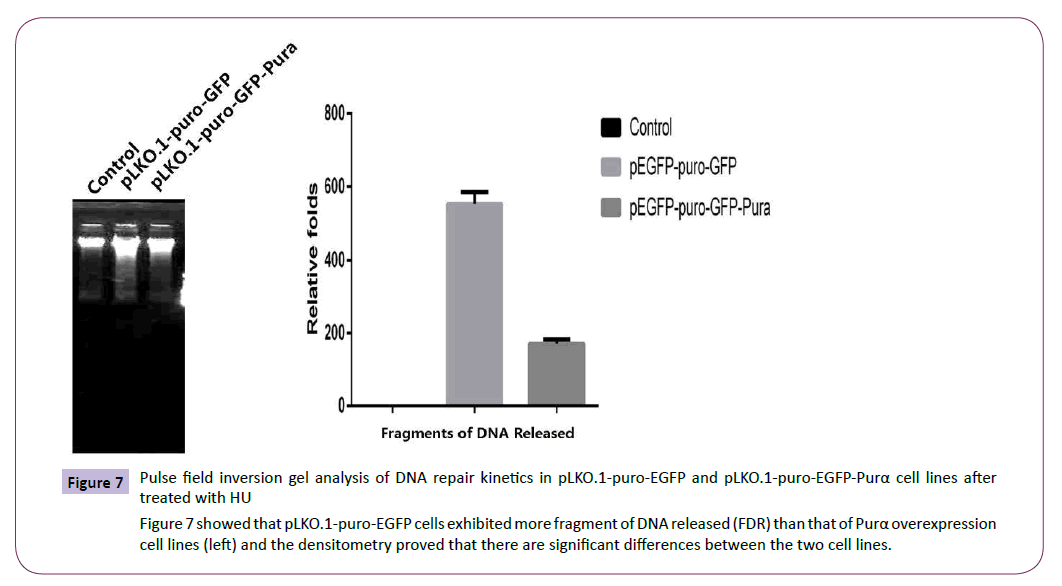
Figure 7: Pulse field inversion gel analysis of DNA repair kinetics in pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα cell lines after treated with HU
Figure 7 showed that pLKO.1-puro-EGFP cells exhibited more fragment of DNA released (FDR) than that of Purα overexpression cell lines (left) and the densitometry proved that there are significant differences between the two cell lines.
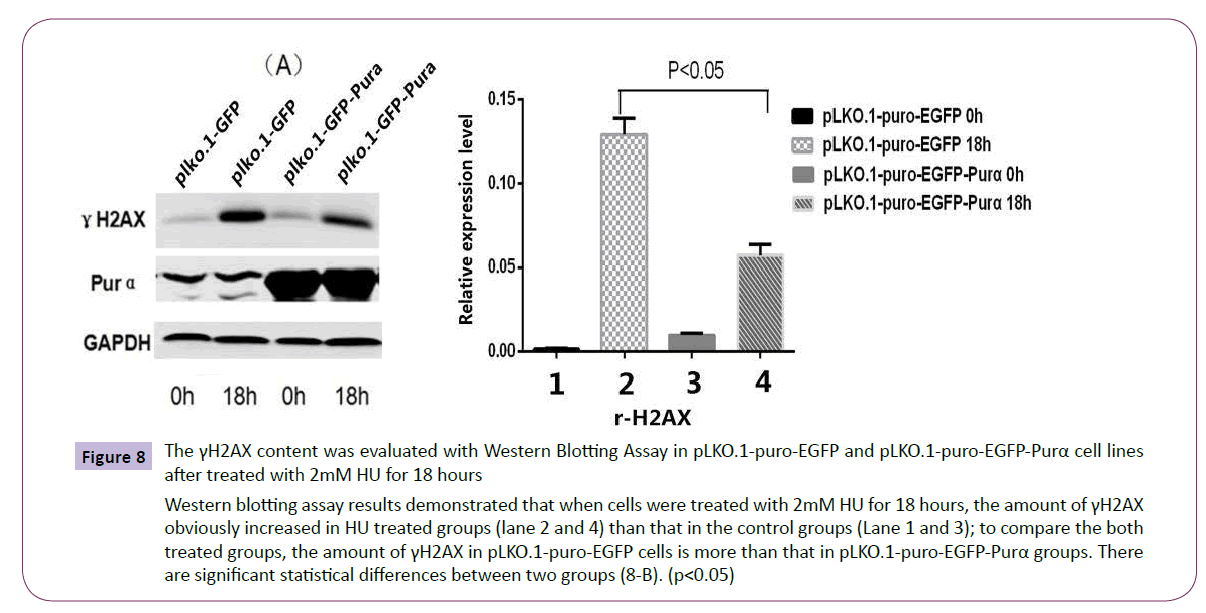
Figure 8: The γH2AX content was evaluated with Western Blotting Assay in pLKO.1-puro-EGFP and pLKO.1-puro-EGFP-Purα cell lines after treated with 2mM HU for 18 hours
Western blotting assay results demonstrated that when cells were treated with 2mM HU for 18 hours, the amount of γH2AX obviously increased in HU treated groups (lane 2 and 4) than that in the control groups (Lane 1 and 3); to compare the both treated groups, the amount of γH2AX in pLKO.1-puro-EGFP cells is more than that in pLKO.1-puro-EGFP-Purα groups. There are significant statistical differences between two groups (8-B). (p<0.05)
Discussion
Purα is a multifunctional protein that binds to single stranded nucleic acid in a sequence specific manner and is a protein family strongly conserved from bacteria throughout humans [8]. Purα has been most extensively characterized as a sequence-specific single stranded DNA and RNA-binding protein originally cloned based on its affinity for a single-strand DNA elements with the sequence GGNGGN [3,4]. Purα plays multiple roles in DNA replication and gene transcription [7,8,15,16], and increasingly recognized as a critical component in the compartmentalized translation of RNA [17]. Purα is a wildly expressed protein, however, it exhibits developmental and tissue-specific regulation [1,11]. For example, Purα protein levels remain quite low in brain during embryogenesis but quickly begin to rise after the first postnatal week in the mouse with levels reaching their peak at 18 days postnatal. Purα then maintains at relatively high levels in the adult brain. Moreover, except its functions in the nucleus, Purα is also strongly detected in the cytoplasm of neurons, particularly in dendrites at branch points and has been found in complex with polyribosomes and heterogeneous nuclear ribonucleoproteins (hnRNP) [18]. Purα also plays an important role in the maintenance of stability of genomic DNA, and specifically expressed in the specific tissues. In central nervous system, it has been approved that that Purα mainly participated in the specific mechanisms associated with transport and translation of mRNA [19,20]. The other recent researches also demonstrated that the mutation of Purα caused a profound neonatal hypotonia, seizures and encephalopathy in 5q31.3 microdeletion syndrome [21],Hunt D. et al with whole exome sequencing in family trios revealed de novo mutations in PURA as a cause of severe neurodevelopmental delay, learning disability, neonatal hypotonia, feeding difficulties, abnormal movements and epilepsy in humans and help clarify the role of PURA in the previous described 5q31.3 microdeletion phenotype [22]. Furthermore, it also has been reported that Purα is associated with endoplasmic reticulum stress and cell differentiation in patients with prostatic cancer [23]. Being a nucleic transcriptional factor, Purα also can bind with RNA particles of neurite cells and inhibit the protein translation [1].
Since Purα knock out genetic mouse die at the 4th week after the birth, it hinder the efforts in the research of function of Purα [11]. The primary cultured mouse embryo fibroblast (MEFs) from Purα knock out mice (Purα-/-) and their wild type mice (Purα+/+) has been successfully established which provided a useful model for the research of Purα functions, but MEFs is not originated from nervous tissue and it is limited in the application in the research in the nervous system [24,25]. In this research we established lentivirus plasmids, pLKO.1-puro-EGFP and pLKO.1-puro-EGFPPurα, which can be wildly used in the research of cells originated from nervous system so that to overcome the low efficiency of the transfection with cells originated from nervous system by chemical method, this model provides a useful tools for the research of Purα in many disciplines. In addition, the constructed lentivirus plasmids were transfected into human glioblastoma cells, U87MG, take advantage of puromycin resistance carried by this vector, the transfected cells were selected with puromycin and successfully established the stable cell lines, pLKO.1-puro- EGFP and pLKO.1-puro-EGFP-Purα, observed under fluorescent microscope which can stably express EGFP and EGFP-Purα proteins. Molecular assays also approved that Purα can be stably and efficiently expressed in this cell lines. Establishment of these cell lines not only meets the requirement for us to deeply investigate the roles Purα played in the cells, but also broaden the requirements for the various relative experiments. The established cell lines express EGFP proteins which can be directly observed under fluorescent microscope, it makes it even more convenient and direct for relevant experiments associated with the functions of Purα in nervous system.
A series experiments were employed to investigate the functions of Purα in the established cell lines. When cells were treated with HU, through CCK-8 examination, we found that as the concentration of HU increased in the treated cells, the proliferation rate became slower; to compare the susceptibility of two cell lines to HU, pLKO.1-puro-EGFP cell lines exhibit more sensitive to HU than that of pLKO.1-puro-EGFP-Purα cell lines, it indicates that overexpression of Purα protein can protect the cells from the cytotoxicity produced by HU. Under normal cell culture condition, the cell growth rate of pLKO.1-puro-EGFP cell lines is faster than that of pLKO.1-puro-EGFP-Purα cell line (data not shown), this implies that in some way, Purα could inhibit the cell growth. This findings is consistent with the reports of Barr SM. et al, when expressed in Ras transformed NIH3T3 cells, Purα can inhibit their ability to grow in the soft agar [26];ectopic expression of Purα suppresses the growth of several tumor cell lines including glioblastoma-origin cells [27]. The growth inhibitory effects of Purα are also supported by the observation that gene expression in chronic myeloid leukemia patients is down regulated by Purα. In Purα knock out mouse embryo fibroblast cell lines (Purα-/- MEFs), cells exhibit a fast growth rate and display characteristics of immortalized cells phenotype, and also prone to cytotoxicity. Re-transfection of Purα gene back into Purα knock out cells can repress the observed phenotypes and restore the normal cell growth [25]. Thus it can be seen that Purα is an important transcriptional factors which plays the critical effects in cell cycle modulation, damaged DNA repair and control of cell proliferation and differentiation.
Neurodegeneration is the common feature of many diseases of the central nervous system, such as Ataxia Telangiectasia, Nijmegen break syndrome, Huntington disease, Parkinson’s disease and Alzheimer’s disease. Furthermore, deficiency in repair of nuclear and mitochondrial DNA damage has been linked to several neurodegenerative disease. This indicates that to maintain the stability of genomic DNA is most important for nervous system [28-30]. The diseases associated with DNA damage can be caused by gene mutation, and all of these genes are associated with the response to DNA damage, some of them are essential for the normal development of nervous system. After the normal developmental process is completed, the neuron enters a post-mitotic state and stay in a resting status. Since lack of the relevant cell cycle proteins and factors, reentry of post-mitotic neuron into cell cycle could not complete the responsible functions for cell cycles, the final consequences will be apoptosis or death. Unrepaired DNA damage promote the neuro reentry into cell cycle to cause the apoptosis and death of the neuron, and also induced the neurodegeneration [31-33]. All these observed phenomena indicate that DNA repair plays an important roles in the development and maintenance of nervous system [28,29]. Therefore, information in repair activities of cells of the nervous system appears necessary for our mechanistic understanding of several related diseases.
HU is usually used for cell synchronization, its effects is reversal. The cells treated with HU will affect the cell’s status in the cell cycle, but when HU is removed, the cell cycle will continue to progress. HU is commonly used as the agents that can cause DNA damage. H2AX is phosphorylated by DNA-PK, ATM and ATR to form phosphorylated H2AX, usually named as γH2AX, and then respond to double stranded breakage of DNA [34-36]. Through pulse field gel analysis, the current research illustrated that when cells were treated with HU, compared with untreated cells, pLKO.1- puro-EGFP cells exhibited the remarkable smears, it suggested that the cells encountered a severe damage and released a great amount of fragments of genomic DNA, but in the pLKO.1-puro- EGFP-Purα cells, there is less smear phenomenon. The results of western blotting assay demonstrated that compared with untreated cells (0 hour), the content of γH2AX is much more in HU treated groups (18 hours). To compare both cells treated with HU for 18 hours, the contents of γH2AX in pLKO.1-puro-EGFP cell group is more than that in pLKO.1-puro-EGFP-Purα cell groups. These results indicate that HU promoted the DNA damage in the cells, but in the pLKO.1-puro-EGFP-Purα cells, the overexpression of Purα alleviated the DMA damage caused by HU and protected the cells from this kind of damage. These results also prompt that Purα might participated in the repair of damaged DNA so that to protect the cell from the damage caused by HU. Wang H. et al has reported that Purα-/- MEFs is much more sensitive to DNA damage reagent HU than that of their counterpart Purα+/+ MEFs, when Purα was re-transfected back into Purα-/- MEFs, the damages caused by HU will be remarkably reversed [25]. The current research work also demonstrated that in our established Purα overexpression cell lines, the damage caused by HU is obviously alleviated, these results confirmed the role of Purα played in DNA repair.
DNA damage and repair is closed associated with neurodegenerative diseases. The unrepaired DNA damage or deficiency of DNA repair will cause the instability of genomic DNA in nervous system, this is the reason for most of the aging disease and degenerative diseases in nervous system [37-39]. Purα protein is closely associated with the neuronal development, synaptogenesis and neurite arborization. In addition, Purα also plays an important roles in the maintenance of stability of genomic DNA in nervous system, participates in DNA repair as well as control and regulate cell cycles [15]. In this sense, it is necessary to investigate the functions of Purα in nervous system, especially the roles it plays in DNA repair and maintenance of the stability of genomic DNA in nervous system. The model we established here will provide a useful approach to further deeply investigate the functions of Purα in nervous system.
Acknowledgements
This work was supported by a Chinese National scientific foundation grant to Dr. Jianqi Cui (81260197) and the Chinese National 973 project grants to Dr. Tao Sun (2012CB722408) and Dr. Jianguo Niu (2014CB560711).
7177
References
- Haas S, Gordon J, Khalili K (1993) A developmentally regulated DNA-binding protein from mouse brain stimulates myelin basic protein gene expression. Mol Cell Biol 13: 3103-3112.
- Haas S, Thatikunta P, Steplewski A, Johnson EM, Khalili K, et al. (1995) A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J Cell Biol 130: 1171-1179.
- Bergemann AD, Johnson EM (1992) The HeLaPur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication. Mol Cell Biol 12: 1257-1265.
- Bergemann AD, Ma ZW, Johnson EM (1992) Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol Cell Biol 12: 5673-5682.
- Ma ZW, Bergemann AD, Johnson EM (1994) Conservation in human and mouse Pur alpha of a motif common to several proteins involved in initiation of DNA replication. Gene 149: 311-314.
- Liu H, Johnson EM (2002) Distinct proteins encoded by alternative transcripts of the PURG gene, located contrapodal to WRN on chromosome 8, determined by differential termination/polyadenylation. Nucleic Acids Res 30: 2417-2426.
- Johnson EM (2003) The Pur protein family: clues to function from recent studies on cancer and AIDS. Anticancer Res 23: 2093-2100.
- Gallia GL, Johnson EM, Khalili K (2000) Puralpha: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res 28: 3197-3205.
- Wang H, Perrault AR, Takeda Y, Qin W, Iliakis G (2003) Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res 31: 5377-5388.
- Deans B, Griffin CS, Maconochie M, Thacker J (2000) Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. EMBO J 19: 6675-6685.
- Khalili K, Del Valle L, Muralidharan V, Gault WJ, Darbinian N, et al. (2003) Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol 23: 6857-6875.
- Crowe SL, Tsukerman S, Gale K, Jorgensen TJ, Kondratyev AD (2011) Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J Neurosci 31: 7648-7656.
- Wang H (2001) Nonhomologous End-Joining of Ionizing Radiation-induced DNA Double-Stranded Breaks in Human Tumor Cells Deficient in BRCA1 or BRCA2. Cancer research 270.
- Wang H, Zeng ZC, Bui TA, DiBiase SJ, Qin W, et al. (2001) Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA1 or BRCA2. Cancer Res 61: 270-277.
- White MK, Johnson EM, Khalili K (2009) Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle 8: 1-7.
- Johnson EM, Daniel DC, Gordon J (2013) Thepur protein family: genetic and structural features in development and disease. J Cell Physiol 228: 930-937.
- Johnson EM, Kinoshita Y, Weinreb DB, Wortman MJ, Simon R, et al. (2006) Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res 83: 929-943.
- Kanai Y, Dohmae N, Hirokawa N (2004) Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43: 513-525.
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, et al. (2007) Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55: 556-564.
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, et al. (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55: 565-571.
- Lalani SR, Zhang J, Schaaf CP, Brown CW, Magoulas P, et al. (2014) Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet 95: 579-583.
- Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, et al. (2014) Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet 51: 806-813.
- Sekiguchi J, Ferguson DO, Chen HT, Yang EM, Earle J, et al. (2001) Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. ProcNatlAcadSci U S A 98: 3243-3248.
- Wang H, Wang M, Reiss K, DarbinianSN, Johnson EM, et al. (2007) Evidence for the involvement of Puralpha in response to DNA replication stress. Cancer BiolTher 6: 596-602.
- Wang H, White MK, Kaminski R, Darbinian N, Amini S, et al. (2008) Role of Puralpha in the modulation of homologous recombination-directed DNA repair by HIV-1 Tat. Anticancer Res 28: 1441-1447.
- Barr SM, Johnson EM (2001) Ras-induced colony formation and anchorage-independent growth inhibited by elevated expression of Puralpha in NIH3T3 cells. J Cell Biochem 81: 621-638.
- Darbinian N, Gallia GL, King J, Del Valle L, Johnson EM, et al. (2001) Growth inhibition of glioblastoma cells by human Pur(alpha). J Cell Physiol 189: 334-340.
- Jackson SP, Durocher D (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49: 795-807.
- Caldecott KW (2004) DNA single-strand breaks and neurodegeneration. DNA Repair (Amst) 3: 875-882.
- Kruman II, Wersto RP, CardozoPF, Smilenov L, Chan SL, et al. (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41: 549-561.
- Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, et al. (2001) Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 24: 25-31.
- Liu DX, Greene LA (2001) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32: 425-438.
- Bossy WE, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10 Suppl, S2-9.
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605-611.
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J BiolChem 273: 5858-5868.
- Wang H, Wang M, Bocker W, Iliakis G (2005) Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J Cell Physiol 202: 492-502.
- Madabhushi R, Pan L, Tsai LH (2014) DNA damage and its links to neurodegeneration. Neuron 83: 266-282.
- Rulten SL, Caldecott KW (2013) DNA strand break repair and neurodegeneration. DNA Repair (Amst) 12: 558-567.
- Jeppesen DK, Bohr VA, Stevnsner T (2011) DNA repair deficiency in neurodegeneration. ProgNeurobiol 94: 166-200.

