Keywords
Fyn kinase; 2D-QSAR; pharmacophore; type I inhibitor; type II inhibitor
Introduction
Fyn kinase is a member of the Src family kinases (SFKs). It is localized to the cytoplasmic side of the plasma membrane, where it phosphorylates tyrosine residues of enzymes involved in different signalling pathways and works downstream of several cell-surface receptors. Fyn kinase is involved in several biological functions. It has a role in mitosis, differentiation of natural killer cells, oligodendrocytes, keratinocytes, growth factor and cytokine receptor signalling, platelet activation, and T-cell and B-cell receptors signalling [1].
Fyn is primarily involved in several transduction pathways in the central nervous system (CNS) and in the peripheral immune system [1,2] playing an important role in regulation and functions of T-cell development and activation [3]. Fyn over expression induces morphogenic transformation, alteration of mitogenic signals and stimulation of cell growth and proliferation. Fyn kinase is also known to mediate integrin adhesion and cell-cell interactions. For all these reasons, Fyn is involved in the onset of cancer [4]. Other studies reveal the engagement of Fyn kinase with other Src-family kinases in signal transduction pathways induced by β-amyloid through their binding to macrophages CD36 receptor that promotes inflammatory changes in response to these proteins accumulated in atherosclerosis [5]. Furthermore, oxLDL enhances CD36 receptor association with Fyn kinase which induces the phosphorylation of Src kinases as Lyn, the subsequent activation of the mitogen-activated kinases Junkinase (JNK) 1 and 2 and the development of atherosclerosis plaque [6].
The involvement of Fyn kinase in the development of diseases makes these protein tyrosine kinases as a potential target for the drugs with therapeutic effects.
Fyn kinase is a protein with 59-kDa and 537 amino acids. This kinase is encoded by Fyn gene which is located on chromosome 6q21. Four principal domains constitute the Fyn kinase: SH1 domain contains the kinase activity; SH2 and SH3 domains mediate protein interactions; and SH4 domain which associated with the cell membrane through their myristylation or palmitoylation. SH1 domain extends along of sequence from amino-acid 271 to 524 of Fyn kinase [4].
The DFG-in conformation of Fyn kinase structure has already been resolved by X-ray crystallography [7] but so far, its DFG-out conformation has not been determined yet. The experimental studies of Fyn kinase inhibition reveal the high selective of some type II inhibitors towards Fyn kinase which assumes the presence of a DFG-out conformation [8].
As mentioned by Jianming Zhang et al., the kinase inhibitors have been classified into four categories based on their mode of binding [9].
Type I inhibitors target the ATP binding site making them as ATP competitive inhibitors.
Type II inhibitors are recognized by kinases on DFG-out conformation. They are characterized by the occupation of the hydrophobic pocket adjacent to that of ATP binding. Also, this type of inhibitors extends to reach the Gatekeeper into the adenine pocket and form hydrogen bonds with the hinge residues.
Type II inhibitors include molecules that occupy the allosteric pocket without reaching the adenine binding pocket [9].
Type III inhibitors do not form hydrogen bonds with hinge residues, they satisfied by the occupation of the back pocket which is an allosteric pocket located at the opposite of ATP binding pocket. This class of inhibitors induces conformational changes in the activation loop, forcing the αC helix to adopt an inactive conformation [9].
Type IV inhibitors bind to one of the allosteric sites distant from the ATP binding pocket inducing a conformational change which renders kinase inactive [9].
In recent years, the researchers have successfully developed a significant number of Src kinase inhibitors. Among these inhibitors, there are those that have been tested in the preclinical and clinical phase. For example, Dasatinib (SRC/ABL inhibitor) which has been approved by the FDA is capable to impair cell migration [10] and inhibits FAK and p130CAS phosphorylation in DU145 and LNCaP prostate cancer cell lines. Also, AZD0530 an inhibitor of growth by inducing G1- arrest in 22Rv1, DU145, LAPC-4, LNCaP, and PC3 prostate cancer cell lines [11].
In this work, we identified small molecules as potent and selective inhibitors of Fyn kinase that could be as a drug therapy for pathologies such as cancer.
The present study combines ligand-based and structurebased drug design approaches. In one hand, 2D quantitative structure-activity relationship (2D-QSAR) was used to generate models from 71 compounds belonging to type I and type II inhibitors and separated to training and test sets. Validation of the QSAR models was judged by root-mean-square error (RMSE) and the correlation factor (R²). In the other hand, pharmacophore modelling was used to select hits for the most active compounds and docking studies of the active inhibitors were carried to determine their binding mode into the active site of Fyn kinase and to interpret the efficacy of the molecules to inhibit Fyn kinase in the lack of crystallized structure of Fyn kinase on DFG-out conformation. This last step relied on the homology modelling to predict 3D-structure of Fyn kinase on DFG-out conformation.
Materials and Methods
2D-QSAR study
The 2D-QSAR models were built via MOE QSAR module [9]. The compounds used in this study belong to the class of type I and type II. The inhibitors of Fyn kinase were extracted from CanSAR database [10] and filtered by Molecular Weight (MW was between 200 Da and 600 Da) and the activity type. The selected activity was IC50 (the concentration of the ligand triggering 50% of the enzyme inhibition). In this step we have obtained 1351compounds. Thereafter, we have selected only molecules which contain 3D structure and the experimental biological activities IC50 value. Then, we have chosen compounds belonging to type I inhibitors and type II inhibitors. A series of 68 compounds was collected. Among these compounds we have selected 24 molecules of type II inhibitors class to which we have added three in house inhibitors of Fyn kinase to create ‘group 1’.
The molecules of ‘group 2’ belongs to the type I inhibitors class that bind to the hinge of the kinase by competing with the ATP molecule. This group contains 44 molecules.
Given the importance of the validation using external method, these groups were divided into training and test set in order to evaluate the QSAR model on a test set of compounds.
The training set of the first group with 23 compounds devote for constructing a 2D-QSAR model and the test set with 4 compounds were used for evaluating the model by prediction of their inhibitory activities. The second group was divided into 38 molecules for training set and 6 molecules for test set.
QuaSAR descriptor module of MOE was used to calculate descriptors for each molecule. 46 molecular descriptors including 2D and 3D descriptors were selected and calculated for the submitted structures aiming to cover a wide range of molecular characteristics. 2D molecular descriptors include physical properties, subdivided surface areas, atom and bond counts, partial charge, pharmacophore feature, Kier & Hall connectivity, kappa shape indices, adjacency and distance matrix.
3D molecular descriptors are classified as ‘i3D’ for internal coordinate dependent 3D and ‘x3D’ for external coordinate dependent. This class includes potential energy, conformation dependent charge, surface area, volume and shape descriptors [11].
The biological activity values IC50 were converted to negative logarithmic (pIC50) and used as the dependent variable for the QSAR analysis.
QSAR models were then constructed after ensuring reasonable correlation of activity with the individual descriptors and minimum inter-correlation among the descriptors used in the derived model.
The quality of the models was assessed using the statistical parameters (correlation coefficient ‘R’, determination coefficient ‘R²’, and Root Mean Square Error ‘RMSE’). The R and R² value should be>0.6 and RMSE should be low<0.3 [12].
The Z-Score value and the cross-validation procedure were used in the process of QSAR model internal validation.
A graphical analysis based on the Z-Score value was used to pinpoint molecules which are outliers to the fit. The crossvalidation evaluates the predicted activities and the residuals for the molecules in the training set. In cross-validation, the model is tested using a Leave One Out ‘LOO’ method.
Excellent values of R² and RMSE are not sufficient indicators of model validity. In order to confirm the reliability of the QSAR models, we have estimated the predicted activities in the test sets.
Pharmacophore modelling
The pharmacophore models were created using MOE pharmacophore module [13]. In this step, we have selected the most active compound on type II and type I inhibitors classes for studying their features. Furthermore, two models were generated to predict the common features among the active molecules presenting the key elements in the binding with the Fyn kinase active site.
The pharmacophore model ‘A’ created on the basis of ‘OR0356’ compound belongs to type II inhibitors with an IC50 of 2.49 nM. The pharmacophore model ‘B’ based on Dasatinib compound (ID:3062316) which have an IC50 equal to 0.2 nM against Fyn kinase. It belongs to type I inhibitors class. Then, the most active molecule was aligned with two other molecules which have a high activity and belongs to the hits of the pharmacophore models using MOE flexible alignment [14]. This step aims to identify common features between the most active molecules of the same class.
To achieve this purpose, we have aligned three molecules of the same class. The first alignment was between ‘OR0356’ compound and two molecules ‘16118737’ and ‘44320732’ with IC50 of 3 nM and 4 nM respectively. The second alignment was between ‘3062316’, ‘56649281’ and ‘11719421’ compounds. IC50 of the last two molecules was 11.9 nM and 23 nM respectively.
Homology modelling
For study the interaction with type II inhibitors and Fyn kinase on DFG-out we have predicted the 3D-structure of Fyn kinase on DFG-out conformation using Modeller 9.11 platform [15]. The Src kinase was selected as the template for prediction by homology modelling method. PDB ID:2OIQ is the crystallized structure of Src kinase in complex with Imatinib that used in this study [16]. The similarity between the two kinases is strong and exceeds 83%. The predicted model of Fyn kinase was validated using the Procheck [17], Errat [18] and Verify 3D [19].
Fyn kinase domain on DFG-in conformation was already crystallized in complex with staurosporine by X-Ray diffraction with a resolution of 2.8Å; their ID in PDB database is 2DQ7 [9].
To verify the Fyn kinase conformations, we have analysed D1, D2 and D3 distances. D1 is the distance between Phe (F) of DFG motif and Asn (N) of HRDXXXXN motif. D2 is the distance between Phe (F) of DFG motif and Cα-atom of the conserved Glu from αC helix. D3 is the distance between Asp (D) of DFG motif and Cα-atom of the conserved Glu from αC helix. The D3 distance calculated in order to verify the αC helix conformation. In the active state, the αC helix conformation called αC-in conformation and αC-out conformation in the inactive state [20].
Docking-based virtual screening
The most similar molecules on type II inhibitors class which were ‘OR0356’ and ‘16118737’ and the three molecules of type I inhibitors class were selected to study their interactions with Fyn kinase by molecular docking approach. In this step, we have used Autodock vina software [20] in order to perform rigid and flexible docking.
The rigid docking relies on a rigid 3D structure of Fyn kinase on DFG-out and DFG-in conformations and a flexible ligand. On the other hand, the flexible docking requires a flexible ligand and flexible residues of the Fyn kinase on their two conformations.
During the preparation of the Fyn kinase files (.pdbqt) used for the flexible docking between Fyn-kinase on DFG-out conformation and the selected ligands, we have set the key loops and residues in the flexible mode. It was the residues of activated and catalytic loops, those from the hinge, the P-loop (Glycine-rich loop), conserved Glu (E) of α-helix, conserved Lys of β-brin.
The interaction between Fyn kinase on DFG-in and type I inhibitors relied on the same steps mentioned above with the exception of the residues selected in the flexible mode. In this step, we have made residues of the hinge, the gatekeeper and the P-loop on flexible mode.
The Grid box dimensions used whether in rigid docking or flexible docking were choosing based on the residues involved in the binding site composition of Fyn kinase in their two conformations.
Results
2D-QSAR study was performed in order to find a mathematical correlation between the structure and physicochemical properties of Fyn kinase inhibitors (type I and type II) and their inhibitory activity that was expressed as pIC50 (-log IC50) using MOE software.
Model I showed in Figure 1a exhibited a correlation between the experimentally observed and predicted values of Fyn inhibitors type II, with RMSE=0.04935, R=0.9995 and R²=0.9990.
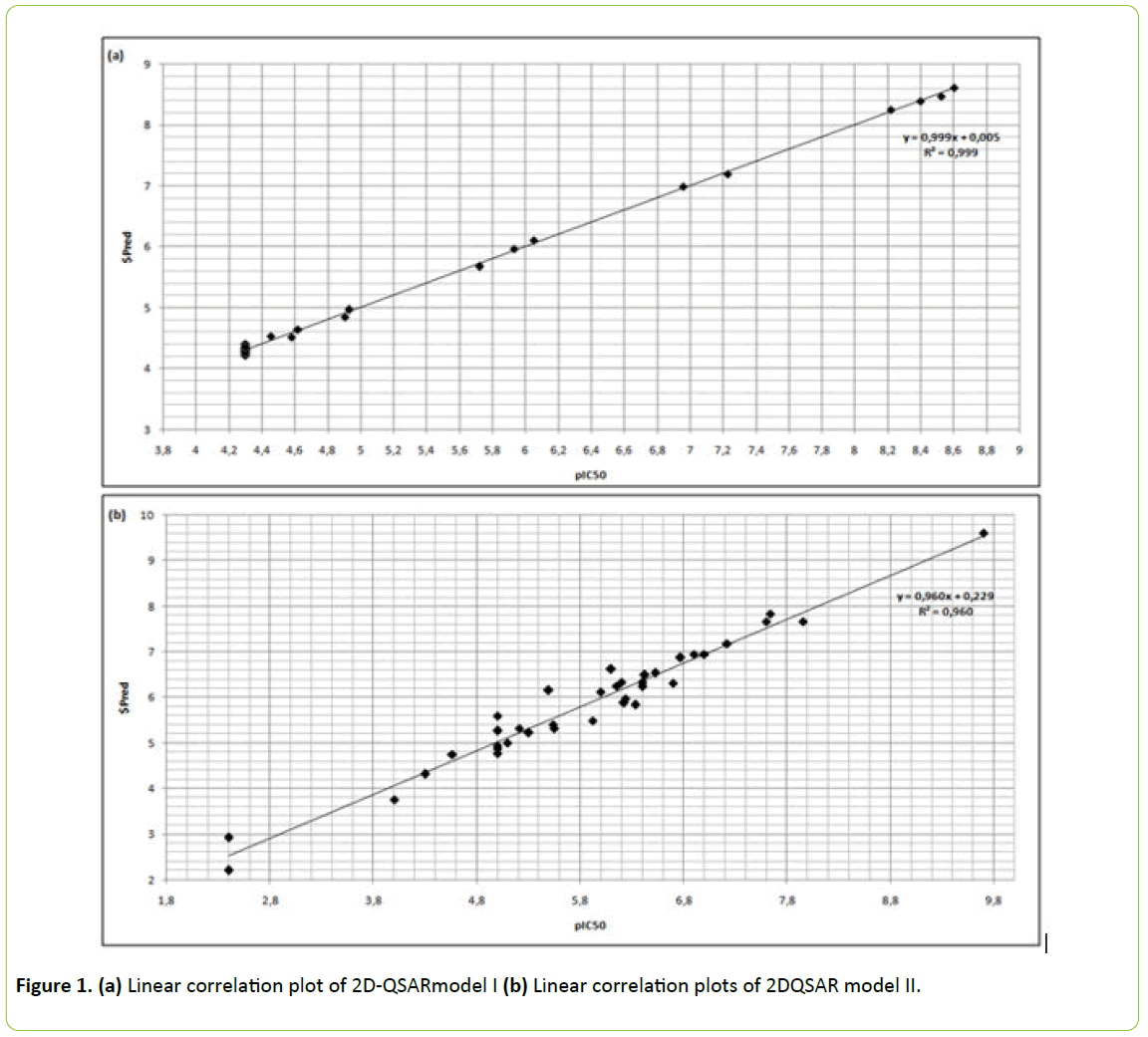
Figure 1: (a) Linear correlation plot of 2D-QSARmodel I (b) Linear correlation plots of 2DQSAR model II.
The resultant correlation regression analysis plot in Figure 1b showed a linear relationship in model II with RMSE=0.27019, R=0.9802 and R²=0.96083.
No molecule was an outlier in the database using MOE’s plotting applications in the two models.
The correlation coefficient between the experimental observed and predicted value of test set compounds of type II inhibitors was 0.78 and 0.71 for test set of type I inhibitors.
The compound ‘OR0670’ in ‘group1’ and ‘10482445’ in ‘group 2’ showed low residual values, which were 0.11 and 0.5247 respectively.
Pharmacophore-based virtual screening
As seen in Figure 2a the pharmacophore model ‘A’ of the most active molecule ‘OR0356’ compound in group 1 reveals 6 hydrogen bond acceptors, 3 hydrogen bond donors, 10 hydrophobic groups and 4 aromatic groups. Hydrophobic groups mainly located around piperazinyl and aromatic groups of the compound.
Figure 2b illustrates the pharmacophore model ‘B’ of Dasatinib. It has 5 hydrogen bond acceptors, 2 hydrogen bond donors, 1 hydrogen bond acceptor/donor, 9 hydrophobic groups and 3 aromatic groups. The hydrophobicity mainly located on the extremity of this compound, precisely around piperazine group and N-(2-chloro-6-methylphenyl).
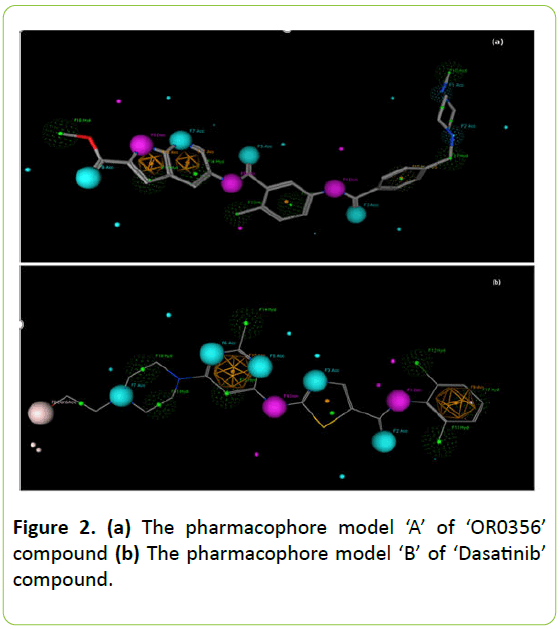
Figure 2: (a) The pharmacophore model ‘A’ of ‘OR0356’ compound (b) The pharmacophore model ‘B’ of ‘Dasatinib’ compound.
The flexible alignment demonstrates the common features between the most active molecules belonging to the same class. The most active molecules of type II inhibitors used in this study have five common radiuses with a score of 100% and five common radiuses by 67% of the score.
Figure 3a exhibits the suggested common features that are within 0.97 angstrom of these molecules. Compounds of type I inhibitors class used in flexible alignment revealed five common radiuses with a score of 100% and three common radius with a score of 67%. Annotations that are within 0.5 are grouped together. Figure 3b shows the design details of their flexible alignment.
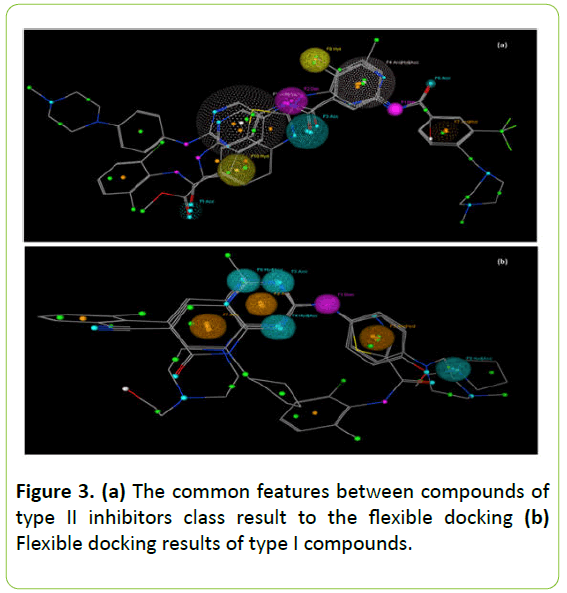
Figure 3: (a) The common features between compounds of type II inhibitors class result to the flexible docking (b) Flexible docking results of type I compounds.
Homology modeling study
100 models were generated by Modeller 9.11 and screened using the lowest DOPE score.
According to Ramachandran plot of Procheck 91.6% of residues are distributed in the most favored regions and no residues in disallowed regions (Figure 4a).
The analytical result score by Verify 3D of predicted Fyn kinase model is low about 55.04% and that of 2OIQ template is 96.99%. The manual verification of Fyn and Src kinases parameters show that the sequence of Fyn kinase amino acids 260-412 is correlated by 79% with the template. That confirms the reliability of our Fyn kinase model.
The global quality factor analysis Errat was 75.188 (Figure 4b) which increases to 90 after elimination of the Fyn kinase downstream part.
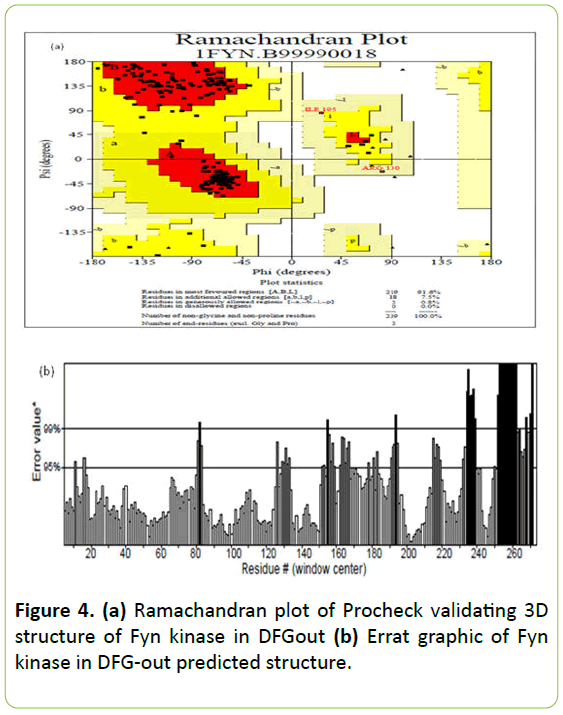
Figure 4: (a) Ramachandran plot of Procheck validating 3D structure of Fyn kinase in DFGout (b) Errat graphic of Fyn kinase in DFG-out predicted structure.
DFG-in active conformation and DFG-out inactive conformation are illustrated in Figure 5a and 5b.
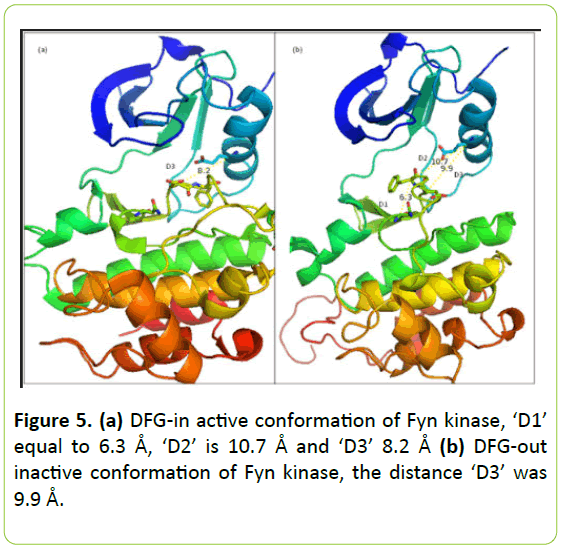
Figure 5: (a) DFG-in active conformation of Fyn kinase, ‘D1’ equal to 6.3 Å, ‘D2’ is 10.7 Å and ‘D3’ 8.2 Å (b) DFG-out inactive conformation of Fyn kinase, the distance ‘D3’ was 9.9 Å.
Like all the members of Src family, it is observed that the 3D structure of Fyn kinase reveals a common bilobal fold consisting of a smaller N-terminal and a larger C-terminal lobe connected by a hinge. The N-lobe contains a five-stranded β sheet and α-helix called the αC-helix, whereas the C-mode is mostly α-helical.
Conformational analysis of the DFG-out of Fyn kinase reveals that ‘D1’ equal to 6.3 Å, ‘D2’ is 10.7 Å and ‘D3’ distance was 9.9 Å in DFG-out conformation of Fyn kinase and 8.2 Å on their DFG-in conformation.
D1 is less than 7.2 Å, D2 is higher than 9 Å and D3 is between 9 Å and 10.5 Å which confirm that the predicted model of DFG-out Fyn kinase is reliable. The downloaded structure of Fyn kinase in DFG-in state is verified by the D3 value. It was less than 9 Å that confirm their active state.
Interaction between Fyn kinase and the compound ‘OR0356’
This OR0356 compound (Methyl5-(5-{p-[(4-methyl-1- piperazinyl) methyl] benzoylamino}toluoylamino)-1,7- diaza-1H-indene-2-carboxylate) was successfully docked into the binding site of Fyn kinase and it was shown to be interacting with the binding site in two ways as is shown in Figure 6a.
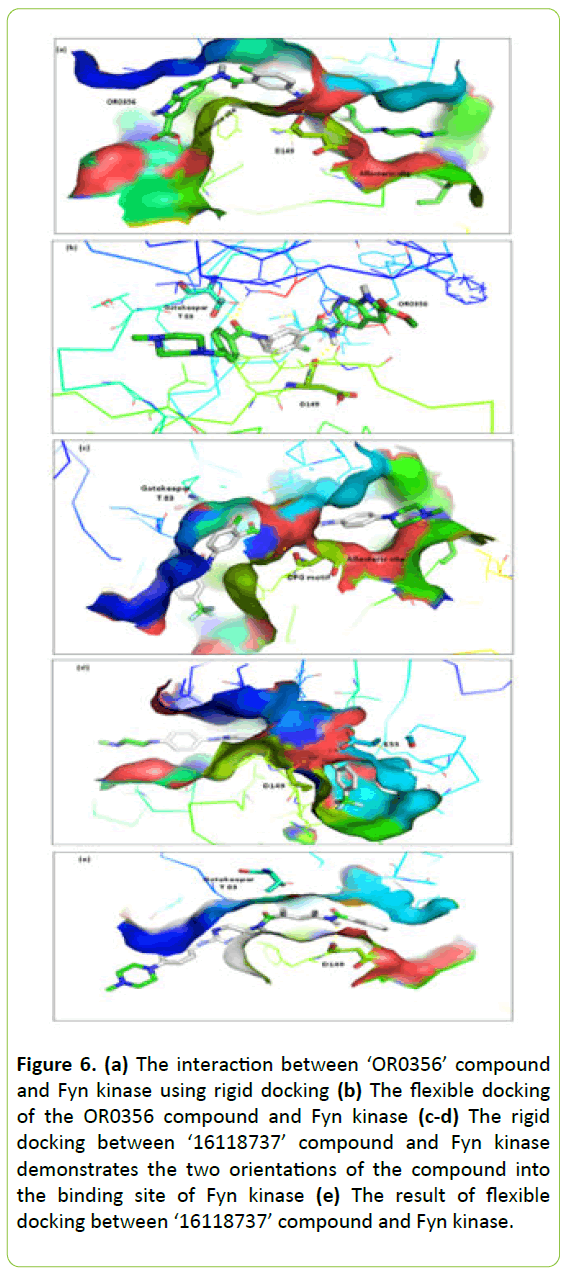
Figure 6: (a) The interaction between ‘OR0356’ compound and Fyn kinase using rigid docking (b) The flexible docking of the OR0356 compound and Fyn kinase (c-d) The rigid docking between ‘16118737’ compound and Fyn kinase demonstrates the two orientations of the compound into the binding site of Fyn kinase (e) The result of flexible docking between ‘16118737’ compound and Fyn kinase.
The rigid docking revealed the interaction between the compound and Fyn kinase using hydrogen bonds and hydrophobic interactions. The compound occupied the allosteric site and the binding site of adenine. In the allosteric site the molecule binds by two hydrogen bonds. The first one is between the amide function of compound and the Asp-149 of DFG motif by their oxygen atom backbone. The second hydrogen bond between Ile-128 just precedes HRD motif in the activation loop and the nitrogen atom of the tail part of the compound. The head part of this compound extends to the adenine region using just the hydrophobic interaction.
The flexible docking of the compound and Fyn kinase in Figure 6b reveals two important hydrogen bonds. The first one with the gatekeeper Thr-83 and the second with the Asp-149 of DFG motif. In this interaction the molecule oriented in a way where the supposed head portion placed in the upstream of the binding site.
Interaction energy between ‘OR0356’ and Fyn kinase during rigid docking was -9.9 kcal/mol and -11.4 kcal/mol using flexible docking.
Interaction between Fyn kinase and the compound ‘16118737’
During the rigid docking, the compound ‘16118737’ (2- methyl-N-[2-[4-(-methylpiperazin-1-yl)anilino]pyrimidin-5- yl]5[[3(trifluoromehtyl)benzoyl]amino]benzamide) oriented into the binding site of Fyn kinase by two distinct ways as illustrated in Figure 6c and 6d).
In the first orientation, their trifluoromethyl group placed on the hydrophobic pocket and methylpiperazine group on the allosteric site. In the second one, the trifluoromethyl group positioned on the allosteric site and the methylpiperazine group on the hydrophobic pocket. The ligand creates two hydrogen bonds; one with the Asp-149 and other with conserved glutamate of Cα-helix Glu-55. This last result of rigid docking was similar to the flexible docking results (Figure 6e). It showed two hydrogen bonds with the key residues, the gatekeeper The-83 and Asp-149 of DFG motif. The supposed tail part of the compound includes trifluoromethyl group placed on the allosteric site by a hydrophobic interaction.
Energy of binding of ‘16118737’ compound was -10.4 kcal/mol and -10.1 kcal/mol during rigid docking and -11.9 kcal/mol during flexible docking.
Preferred orientation into Fyn kinase of ‘3062316’ compound
Dasatinib (N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2- hydroxyethyl) piperazin-1-yl]-2 methylpyrimidin-4-yl] amino]-1,3-thiazole-5-carboxamide) occupied the adenine region and the hydrophobic regions I and II.
Figure 7a shows the interaction results of rigid docking when the binding energy was -8.8 kcal/mol. The interaction between Dasatinib and Fyn kinase using flexible docking reveals their placement in the adenine region and hydrophobic regions by hydrophobic interaction and two hydrogen bonds with Tyr-84 and Asn-86 of the hinge and one bond with Ala-134 (Figure 7b). However, the flexible docking reveals some placement of Dasatinib in the binding site of Fyn kinase to three hydrogen bonds with the gatekeeper The-82 and Met-85 of the hinge with an interaction energy of -8.0 kcal/ mol.
Analysis of interaction between Fyn kinase and ‘56649281’ compound
The ‘56649281’ compound (8-cyclopentyl-2-[4-(4- methylpiperazin-1-yl) anilino]-7-oxopyrido [2,3-d]pyrimidine-6- carbonitrile) occupied the Fyn kinase binding site using hydrogen bonds and hydrophobic interactions. It successfully binds with Met-85 of the hinge by two hydrogen bonds whether using rigid or flexible docking as shown in Figure 7c and 7d). Their binding energy was -9.3 kcal/mol in rigid and flexible mode interaction.
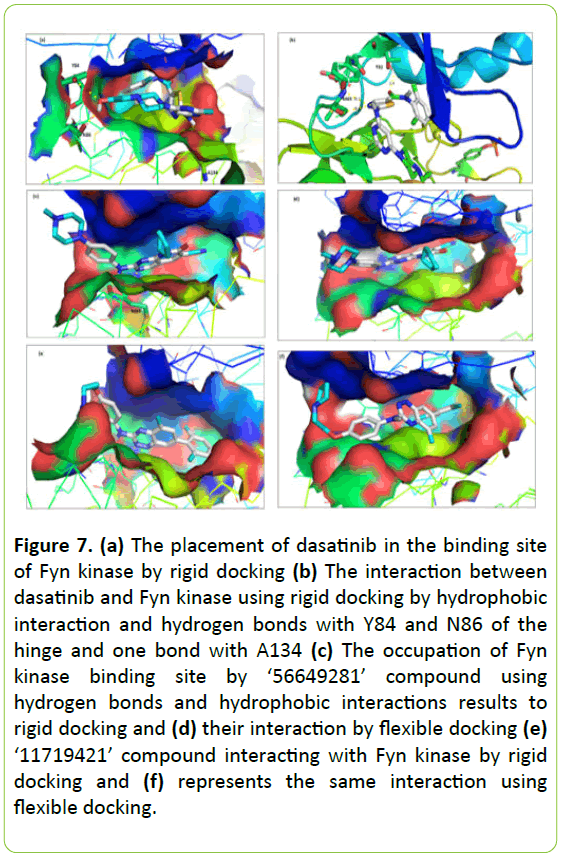
Figure 7: (a) The placement of dasatinib in the binding site of Fyn kinase by rigid docking (b) The interaction between dasatinib and Fyn kinase using rigid docking by hydrophobic interaction and hydrogen bonds with Y84 and N86 of the hinge and one bond with A134 (c) The occupation of Fyn kinase binding site by ‘56649281’ compound using hydrogen bonds and hydrophobic interactions results to rigid docking and (d) their interaction by flexible docking (e) ‘11719421’ compound interacting with Fyn kinase by rigid docking and (f) represents the same interaction using flexible docking.
Predicted interaction between Fyn kinase and ‘11719421’ compound
As shown in Figure 7e and 7f, the rigid and flexible docking demonstrates that the compound ‘11719421’ (7-(2,6- dimethylphenyl)-5-methyl-N-[4-(2-pyrrolidin-1- ylethoxy)phenyl]-1,2,4-benzotriazin-3-amine) positioned into the ATP-binding site of Fyn kinase occupying their sub-sites. It binds with the Met-85 of the hinge by two hydrogen bonds. Their interacting energy in rigid and flexible mode interaction was -9.3 kcal/mol and -9.1 kcal/ mol respectively.
Discussion
In this report, we showed that identified 2D-QSAR models generated are reliable and useful for the activity prediction of new compounds with a scaffold close to molecules used in these models and for study the physicochemical characteristics of molecules.
Molecular descriptors analysis exhibited that eight molecules of ‘group 1’ and 31 molecules of ‘group 2’ were predicted to have superior drug like properties, they satisfied all Lipinski rules of five [21].
The most active inhibitors of type II docked with Fyn kinase satisfied three of the four Lipinski criteria. Their molecular weights are quite high to 500 Da but remain lower to 590 Da. Furthermore, two of the three most active compounds in ‘group 2’ satisfied Lipinski rules.
We have studied the interaction between Fyn kinase on DFG-out conformation and the most active type II inhibitors used in this research. The molecular docking results confirmed the selectivity of Fyn kinase to type II inhibitors with assay concentrations.
It was noted that among the studied molecules some have binding affinity with the allosteric site created by DFG-out conformation. This affinity results to the hydrophobic interaction and hydrogen bonding combination.
The studied compounds have a head group that extends to reach the adenine region using a hydrogen-binding donoracceptor pair and contain a hydrophobic portion (tail) that interacts with the allosteric site.
The docked type II compounds contain trifluoromethyl group. They have an important role in the compound position into the binding pocket of Fyn kinase. We have deduced that trifluoromethyl group can act in the orientation of molecules in the binding site by occupying of the most hydrophobic pocket.
Fluorine is significantly more lipophilic than hydrogen and its incorporation into a molecule will make it more liposoluble. The liposolibility of fluorinated molecule increases their bioavailability. Indeed, fluorine has become a very interesting tool in drug discovery [22].
It was observed that piperazine group is frequently incorporated in the design of type II inhibitors in order to occupy one of the hydrophobic pockets in the binding site of enzymes.
Type II inhibitors are capable to create a pair of hydrogen bond with the allosteric site residues using an amide or urea. One of this bond pair is with the glutamic acid side chain from αC-helix and the other is with the Asp of DFG motif.
In addition, this type of inhibitor interacts with the allosteric site by their hydrophobic part through Van der Waals interaction. Also, it can extend into the adenine region forming one or two hydrogen bonds with the hinge residues.
In comparing which has already been recognized about the binding mode of type II inhibitors with the docking results of this study we concluded that the two docked compounds are successfully placed into the binding site of Fyn kinase.
The noticed orientation of the studied compound in the binding pocket of Fyn kinase results to the presence of two amide functions, and hydrophobic extremities.
The type I kinase inhibitors binding mechanism is already explained by P. Traxler and P. Furet in the paper of ‘Strategies toward the Design of Novel and Selective Protein Tyrosine Kinase Inhibitor’.
This type of inhibitor does not require DFG-out conformation for binding. It is satisfied by the occupation of the area reserved for the adenine ring of ATP building hydrogen bonds with the kinase hinge residues by mimicking the normal ones formed by the exocyclic amino group of adenines [22].
Among studied type I compounds some contain aromatic heterocyclic group which targets the hydrophobic pockets and try to create bond with the hinge by their nitrogen atoms.
The molecular dynamics study of Fyn kinase crystal structure in the absence of inhibitor showed the limitation of ATP binding which results in flexibility of the glycine-rich loop [10]. This result allowed recommending by the necessity to consider this flexibility in the design of new inhibitors.
The interaction between Dasatinib and Fyn kinase using rigid docking reveals their placement in the adenine region and hydrophobic regions by hydrophobic interaction in the absence of hydrogen bonds. However, the flexible docking reveals some placement of Dasatinib in the binding site of Fyn kinase using 2 to 3 hydrogen bonds with the hinge.
The crystallized Dasatinib in complex with Lyn, Src and Abl2/Arg kinases showed that Dasatinib binds with the hinge of these kinases by three hydrogen bonds [22-24]. This interaction mode is similar to the obtained flexible docking results of Dasatinib in interacting with Fyn kinase in our work.
Our study shows that the most active Fyn kinase inhibitory molecule «OR0356 compound» can be a lead one and need to be biologically analysed. Therefore, we suggest in the future investigating further this molecule by measuring their pharmacokinetic properties such as absorption, distribution, metabolism and elimination.
Conclusion
We have performed a structure-activity relationships study of Fyn kinase as potential therapeutic targets because of its involvement in a multitude of physiological processes.
QSAR study of Fyn kinase inhibitors has given us an idea of the physicochemical features of studied compounds and their correlation with the IC50 activity. Note that it is interesting to identify the physicochemical proprieties of a new therapeutically active molecule. Furthermore, the docking and pharmacophore studies have helped to highlight the molecule key elements to refine in order to get a selective and more potent compound of Fyn kinase.
Type I inhibitors have the ability to bind to the ATP site, therefore this binding is not specific due to the fact that these components can be recognized by other members of the kinase target family. The clinical side effects are often severe with this lack of specificity.
However, we have studied type I inhibitors of Fyn kinase for two reasons. The first one was to verify the placement of these inhibitors into Fyn kinase on DFG-in, due to the lack of a crystallized complex (except for Fyn kinase-staurosporine). The second reason was to create an extensive database of Fyn kinase type I inhibitors to serve as the basis for developing type II inhibitors which can be broken down into two portions.
The deep study of type II inhibitors will remain useful to interpret their interaction mode with Fyn kinase on DFG-out despite the lack of its crystallized structure.
Acknowledgement
The authors thank the participants and their relatives and the staff of FraDySMex study.
23281
References
- Cooke MP (1991) Regulation of T Cell Receptor Signaling by a Src Family Protein-Tyrosine Kinase (p59fyn). 65: 281-291.
- Sperber BR and McMorris FA (2001 ) Fyn Tyrosine Kinase Regulates Oligodendroglial Cell Development but Is Not Required for Morphological Differentiation of Oligodendrocytes. Journal of Neuroscience Research 63: 303-312.
- Palacios EH and Arthur Weiss (2004 ) Function of the Src-Family Kinases, Lck and Fyn, in T-Cell Development and Activation. Oncogene 23: 7990-8000.
- Kawakami T (1988) Acquisition of Transforming Properties by FYN, a Normal SRC-Related Human Gene. Proceedings of the National Academy of Sciences of the United States of America 85: 3870-3874.
- Moore KJ (2002) ) A CD36-Initiated Signaling Cascade Mediates Inflammatory Effects of β-Amyloid. Journal of Biological Chemistry 277: 47373-47379.
- Samovski D (2015) Regulation of AMPK Activation by CD36 Links Fatty Acid Uptake to β-Oxidation. Diabetes 64: 353-359.
- Kinoshita T (2006) Structure of Human Fyn Kinase Domain Complexed with Staurosporine. Biochemical and Biophysical Research Communications, 346: 840-844.
- Schenone S (2011) Fyn Kinase in Brain Diseases and Cancer: The Search for Inhibitors. Current Medicinal Chemistry 18:2921-2942.
- Tym JE (2016) canSAR: An Updated Cancer Research and Drug Discovery Knowledgebase. Nucleic Acids Research 44: D938-D943.
- Gramatica P (2007) Principles of QSAR Models Validation: Internal and External. QSAR & Combinatorial Science 26: 694-701.
- Labute P (2001) Flexible Alignment of Small Molecules. Journal of Medicinal Chemistry 44: 1483-1490.
- Sali A and Blundell TL (1993) Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology 234: 779-815.
- Seeliger MA (2007) C-Src Binds to the Cancer Drug Imatinib with an Inactive Abl/c-Kit Conformation and a Distributed Thermodynamic Penalty 15: 299-311.
- Colovos C and Yeates TO (1993) Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Science: A Publication of the Protein Society 2: 1511-1519.
- Eisenberg D, Lüthy R and Bowie JU (1997) VERIFY3D: Assessment of Protein Models with Three-Dimensional Profiles. Methods in Enzymology 277: 396-404.
- Trott O and Olson AJ (2010) AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry 31: 455-461.
- Lipinski CA (2001) Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews 46: 3-26.
- Ha BH (2015) Structure of the ABL2/ARG Kinase in Complex with Dasatinib. Acta Crystallographica. Section F, Structural Biology Communications 71: 443-448.
- Williams NK (2009) Crystal Structures of the Lyn Protein Tyrosine Kinase Domain in Its Apo- and Inhibitor-Bound State. The Journal of Biological Chemistry 284: 284-291.
- Getlik M (2009) Hybrid Compound Design to Overcome the Gatekeeper T338M Mutation in cSrc. Journal of Medicinal Chemistry 52: 3915-3926.