Keywords
Excitotoxicity; Glutamate; NMDA receptors; Memantine; Cell death
Introduction
Excitotoxicity is a form of neuronal death caused by hyperactivity of excitatory amino acids –mainly Glutamate (Glu)-in the mammal Central Nervous System (CNS) [1]. This activity leads to an excessive cellular influx of ions, particularly calcium, causing the activation of proteases, phospholipases and endonucleases [2].
The neurotoxic effects of Glu were first identified in reports by Lucas and Newhouse in 1957 [3], with studies in mouse retina samples in which after parenteral administration of L-Glutamate, damage to the inner layers of the retina was observed, manifested as partial necrosis of the ganglionic and inner nuclear cells. In the 1960s, further studies emerged on this neurotransmitter and others, such as γ-aminobutyric acid (GABA). The term “excitotoxin” was coined to designate excitatory amino acids with neurotoxic effects [4]; and Glu was discovered to be involved in calcium release after cell depolarization [5]. Later, during the 1970s, the glutamate-glutamine cycle was first described, and Glu was found to be reabsorbed by glia cells following its release to the synaptic cleft. Here, it is converted back to glutamine which returns to the synaptic terminals, where is converted back to Glu by the glutaminase enzyme [6].
Excitotoxicity is a complex process that has been associated with an important number of pathologic conditions with great clinical relevance. These include hypoxic-ischemic states [7], hypoglycemia [8], status epilepticus [9], neurodegenerative diseases [1], head trauma [10], as well as with multiple other neuropsychiatric disorders, such as substance abuse and withdrawal disorders, major depression and schizophrenia [11-13]. In view of the prominent role excitotoxicity plays in these conditions, and the potential therapeutic implications of this phenomenon in the management of these disorders, the purpose of this article is to summarize current molecular and neurobiological views on the pathophysiology of excitotoxicity, conceived as a crime or murder of neuronal cells.
The Synapse: Scene of The Crime
Neuronal impulses are transmitted through synapses, the sites where the axon of a presynaptic neuron comes into proximity with a postsynaptic excitable cell. Synapses might be electrical or chemical. In the former, postsynaptic and presynaptic membranes form communicating junctions, which act as high conductance ionic bridges [14]. However, almost all synapses in the human CNS are of chemical nature. In this type of synapses, a molecule released by the presynaptic terminal –also called a neurotransmitter (NT) – mediates the activation of the target cell [15].
Once the NT is released into the synaptic cleft, it may bind to receptors in the postsynaptic membrane. In general, the basic structure of these receptors includes a ligand-binding site and an active site. Ionotropic receptors allow ionic diffusion after being activated by the NT, which causes a change in their structure to a channel-like conformation [16]. Depending on the ion transported through these channels, an ionotropic receptor might be excitatory if it favors the travel of cations, mainly sodium; or inhibitory, if it favors the travel of anions, mainly chloride. Conversely, metabotropic receptors initiate diverse intracellular signaling cascades through second messengers, which in turn trigger an excitatory or inhibitory net effect [17]. Indeed, the structural and functional characteristics of the postsynaptic receptors determine the excitatory or inhibitory nature of the synapses. Figure 1 illustrates this process.
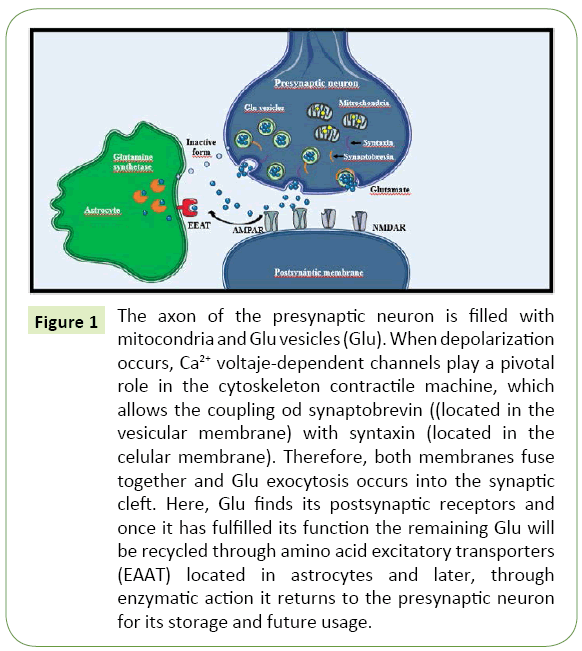
Figure 1: The axon of the presynaptic neuron is filled with mitocondria and Glu vesicles (Glu). When depolarization occurs, Ca2+ voltaje-dependent channels play a pivotal role in the cytoskeleton contractile machine, which allows the coupling od synaptobrevin ((located in the vesicular membrane) with syntaxin (located in the celular membrane). Therefore, both membranes fuse together and Glu exocytosis occurs into the synaptic cleft. Here, Glu finds its postsynaptic receptors and once it has fulfilled its function the remaining Glu will be recycled through amino acid excitatory transporters (EAAT) located in astrocytes and later, through enzymatic action it returns to the presynaptic neuron for its storage and future usage.
Activation of postsynaptic receptors results in excitatory or inhibitory modifications to the membrane potential of the target cell; which may be enhanced by spatial or temporal summation. Changes in the membrane potential lead to depolarization or hyperpolarization of the target cell [18]. To this end, the neuron must reach a minus negative voltage of -45 mV from a basal potential of -65 mV in order to be depolarized and activate the voltage dependent calcium channels that will trigger the subsequent synapsis [19].
Glutamate, Receptors and Calcium: The Criminals
Glutamate: Intellectual author
Synthesis of Glu is the first step in the chain of events involved in excitotoxicity [20]. In the CNS, Glu is mainly synthesized from endogenous precursors, chiefly α-ketoglutarate, a metabolite from the Krebs Cycle [21]. However, the majority of Glu utilized in synapses derives from the recycling of this molecule in a dynamic pool maintained through the glutamate-glutamine cycle [22]. In this purport, Excitatory Amino Acid Transporters (EAAT) – subtypes EAAT1 and EAAT2-located in astrocyte membranes, may withdraw Glu from the synaptic cleft, ending synaptic signaling. Once in the cytoplasm of astrocytes, Glu is converted back to glutamine through glutamine synthase, and then released to the extracellular space in this inactive form, which may be recaptured by presynaptic terminals. It is here where glutamine might be reconverted to Glu by mitochondrial glutaminase, effectively returning Glu back to its place of origin, where it can be reutilized as a NT [23].
Therefore, the glutamine-glutamate cycle is essential, as it reduces the requirement of de novo synthesis of Glu from the Krebs Cycle and regulates Glu activity through EAATs, thus favoring neuronal energetic stability [24].
Glu excess may have multiple origins, reflecting the different conditions that share excitotoxicity as a common mechanism. These include energetic homeostasis disruptions secondary to hypoxic-ischemic states and hypoglycemic states in the CNS [25]. These conditions interrupt the supply of oxygen and glucose to the neurons, which results in disruption of oxidative phosphorylation in the mitochondria, reducing ATP levels. Although there are alternative substrates to glucose, such as glycogen, lactate and fatty acids, oxygen is irreplaceable in mitochondrial oxidative phosphorylation. Therefore, hypoxic-ischemic states stimulate glycogen catabolism, leading to the accumulation of protons and lactate, and therefore, to rapid intracellular acidification and an even greater decrease in ATP availability [26].
Eventually, oxidative phosphorylation fails and ATP synthase activity shifts from ATP production to consumption. At critical ATP concentrations, the sodium/potassium pump also fails, causing neuron and astrocyte depolarization [27]. These changes in voltage driven by membrane depolarization and the change in concentration of sodium and potassium result in activation of voltage-dependent calcium channels. This leads to excessive release of excitatory amino acids such as Glu to the extracellular compartment [26]. In addition, in hypoxic-ischemic states, intracellular sodium accumulation drives a shift in the activity of Glu transporters, from moving Glu from the extracellular space into glial cells, to moving Glu from glial cells to the extracellular space, where this NT then accumulates. This results in excitotoxicity [28].
Glutamate receptors: Accomplices
Glu exerts its effects in target cells through ionotropic and metabotropic receptors. The former play a fundamental part in excitotoxicity. These receptors form 3 major groups denominated according to their corresponding selective agonists: 1) N-Methyl-D-Aspartate (NMDAR); 2) α-Amino-3-Hydroxi-Methyl-4- Isoxazolpropionate (AMPAR); and 3) Kainic acid receptors (KAR) [29].
NMDARs are associated with non-selective ionic channels that allow the preferential calcium and sodium transport to the intracellular space, as well as the exit of intracellular potassium (Figure 2). Due to their capability for calcium transport, NMDARs are especially important in excitotoxicity [30]. Allosteric binding of various modulating molecules such as glycine might modify the activity of these receptors. Glycine, which is a necessary co-agonist for their proper function [31]. Hydrogen ions–as a reflection of local pH–may suppress their activation whilst polyamines may potentiate their activity [32].
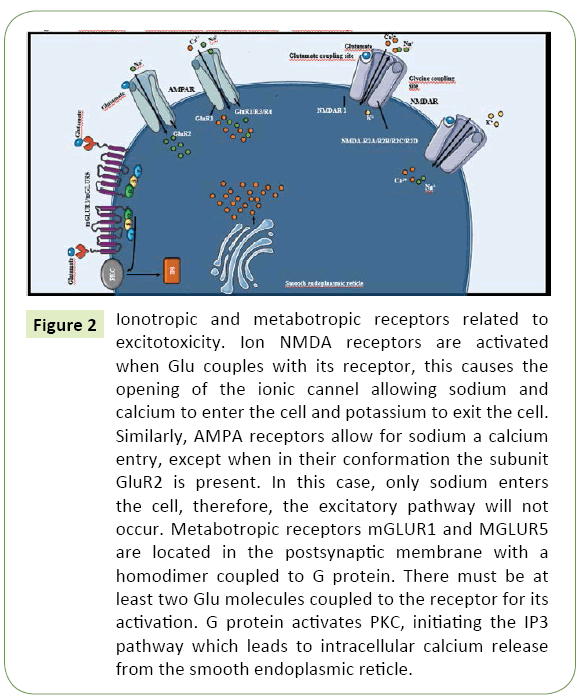
Figure 2: Ionotropic and metabotropic receptors related to excitotoxicity. Ion NMDA receptors are activated when Glu couples with its receptor, this causes the opening of the ionic cannel allowing sodium and calcium to enter the cell and potassium to exit the cell. Similarly, AMPA receptors allow for sodium a calcium entry, except when in their conformation the subunit GluR2 is present. In this case, only sodium enters the cell, therefore, the excitatory pathway will not occur. Metabotropic receptors mGLUR1 and MGLUR5 are located in the postsynaptic membrane with a homodimer coupled to G protein. There must be at least two Glu molecules coupled to the receptor for its activation. G protein activates PKC, initiating the IP3 pathway which leads to intracellular calcium release from the smooth endoplasmic reticle.
On the other hand, AMPAR permeability to calcium is variable and depends on the presence of the GluR2 subunit. Unlike the other subunits which comprise the AMPAR tetramere (GluR1, GluR3 and GluR4), GluR2 is rich in arginine residues. The positive charge of this amino acid blocks calcium transport; thus, transport of cations is restricted to sodium and potassium only [33]. Therefore, only the forms without this subunit contribute significantly to excitotoxicity [34]. In a similar fashion, KAR permeability to calcium also varies according to its structure, specifically the presence of a p loop in its M2 domain [29].
Furthermore, metabotropic Glu receptors are G protein-coupled, and subdivided in three groups according to their specific antagonists: Group I (3,5-dihydroxifenilglycine) which includes variants mGluR1 and mGluR5; Group II (eglumegad, byphenilindanone A and DCG-IV) which includes mGluR2-3 and Group III (L-AP4) that includes mGluR4 and mGluR6-8 [35]. Variants from Group I are located in the postsynaptic membrane and potentiate excitotoxicity as key mediators of the later mechanisms. Through subunit Gq, PLC can be activated, initiating the triphosphate inositol pathway that results in calcium release from intracellular deposits, as observed in Figure 3 [36]. Conversely, group II and III are located in the presynaptic membrane, coupled with subunits Gi/Go. When activated, inhibition of cAMP synthesis occurs. In consequence, these groups act as indirect regulators in postsynaptic Glu receptor activity, augmenting the inhibitory potential of the presynaptic membrane [35].

Figure 3: Cell death mechanisms mediated by excitotoxicity. A) When the glutamate binds to its NMDA receptor, the transport of Na+ and Ca2+ into the postsynaptic neuron is initiated B) There is a change in the intracellular voltage that triggers the opening of Ca2+ channels, while NMDA receptors continue to allow the entry of Ca2+ and Na+ to the cell. The high concentration of Ca2+ alters the permeability of the mitochondria, allowing the entry of this ion. C) Mitochondria trigger cell death processes, mediated by: 1. Apoptosis: activation of Bim, Bax and Bak that activate cytochrome c which in turn participates in the activation of caspase enzymes that catalyze the destruction of genetic material of the cell. 2. Necrosis: cytochrome c activates phospholipase enzymes that act at the membrane level and proteases that act at the cytoskeletal and cellular matrix levels, eventually leading to neuronal death.
Glutamate transporters: The cleaning crew
Glu levels in the extracellular space must be tightly regulated in order to avoid prolonged receptor activation and its subsequent rise to toxic concentrations [37]. To this end, Glu transporters retake Glu from the synaptic cleft only seconds after it is released. These EAATs are Na+ and K+ dependent, and are located in the cellular membrane of glial cells and neurons [38]. Although their physiology is yet to be fully elucidated, EAATs are a family of transporters that present great homology amongst each other, which appear to be the main regulators of extracellular Glu concentrations [39]. The EAAT family includes EAAT1 to EAAT5; all of which catalyze Glu transport through a co-transport with Na+ and K+. Through this mechanism, L-Glutamate, L and D-Aspartate can be transported but not D-Glutamate. It has been established that regulation of its expression is given by different mechanisms in every level, from transcription to posttranslational modifications [40].
Each EAAT isoform shows distinct cellular locations and molecular characteristics: EAAT1 is a Glu and aspartate transporter, whilst EAAT2 transports only Glu, and both may be found throughout all neurons in the SNC, although EAAT1 is mainly located in the cerebellum [41]. These are the only transporters expressed in astrocytes, whilst EAAT3 is expressed only in neurons in different regions of the brain at low levels. EAAT1, EAAT2 and EAAT3 execute their transport function by exchanging an intracellular potassium molecule for a Glu molecule, three sodium ions, and a hydrogen ion, all of which are taken from the extracellular space. They also function as Cl- channels, similar to EAAT4 and EAAT5, which are located only in the brain and retina and have a greater conductance to chloride, working even as Glu inhibitors [42,43].
The most abundant of the transporters is EAAT2, which is expressed mainly in astrocytes, and reabsorbs Glu from the synaptic cleft, being responsible for up to 95% of its total uptake. EAAT2 dysfunction leads to extracellular Glu accumulation, and it has been associated with neurodegenerative diseases like Alzheimer’s Disease (AD), Huntington’s disease and Amyotrophic Multiple Sclerosis [44].
Calcium: Material author
Calcium is the final actor in excitotoxicity, being the main culprit of the direct damaging processes in the neuron. Calcium is a divalent cation known for its prominent role as a messenger in multiple metabolic pathways, acting when its intracellular concentration increases [45]. Therefore, calcium levels are kept at lower intracellular concentrations in comparison to extracellular levels through ATP-coupled membrane transporters: A cation antiporter that interchanges intracellular calcium for extracellular sodium and the calcium plasma-membrane pump [46]. Similarly, mitochondria and the smooth endoplasmic reticulum sequester intracellular calcium, also mediated by active transport systems: An ATP-dependent pump, and an antiporter cation protein in the smooth endoplasmic reticulum [47].
These mechanisms are altered in excitotoxicity, not only by Glu hyperstimulation, but also by the decrease in ATP concentration, which impedes the activity of these active transport systems [48]. In this scenario, the consequent neuronal depolarization results in the opening of voltage-dependent calcium channels, exacerbating the already high intracellular concentration and potentiating its harmful effects [1]. Even though the N and T channels are the more prevalent calcium channel types in the CNS, subtype L –characterized by longer opening time and conductance-plays a fundamental role as mediator of delayed excitotoxic mechanisms [49].
Molecular Mechanisms in Excitotoxicity: Reconstructing the Crime Scene
Excitotoxicity may occur acutely or chronically. Acute excitotoxicity is mediated mainly by the increase of extracellular Glu levels, which leads to an excessive depolarization of the postsynaptic membrane and the subsequent sodium, chloride and water influx into neurons, finalizing in membrane rupture and cellular death [1]. In contrast, chronic excitotoxicity tends to occur in the context of local energy production decreases, where typically non-toxic Glu quantities become deleterious; as seen in the penumbra found after ischemic events [50]. Chronic excitotoxicity may also be sustained by Glu transporters anomalies [51]. In the following paragraphs, we describe the main mechanisms common to both acute and chronic excitotoxicity, and those specific to chronic settings.
Modus Operandi: Mechanisms found in acute and chronic settings
Death by calcium overdose: Enzymatic disaster: In excitotoxicity, massive NMDAR stimulation leads to the loss of ionic homeostasis and excess intracellular calcium [52]. This triggers a series of enzymatic reactions that end in cellular death, which include the following:
Calcium entry: When excess Glu massively stimulates NMDARs, these receptors are permanently activated, causing an excessive sodium and calcium entry to the neuron. This leads to greater influx towards the mitochondria, resulting in mitochondrial dysfunction, with various possible consequences [2]:
Mitochondrial permeability pore: The internal membrane of the mitochondria features a high conductance transition pore. Although its complete structure remains to be fully elucidated, it appears to include a voltage-dependent channel, an adenine translocator, cyclophilin D and a phosphate transporter [53,54]. All of these components have specific contact points between the internal and external mitochondrial membrane [55]. This channel may be activated by excessive calcium influx to the mitochondria or by reactive oxygen species (ROS) [56]. The formation of this pore appears to begin with the translocation of cyclophilin D from the mitochondrial matrix to the internal mitochondrial membrane (IMM), in order to couple with the adenine nucleotide translocator. This connects the mitochondrial matrix with the cytosol through a channel in the IMM, with a voltage-dependent portion in the external mitochondrial membrane (EMM) [57]. Another possible mechanism involved in the formation of the mitochondrial permeability pore involves binding of the phosphate transporter with the adenine translocator [58].
Once formed, influx of ions and water into the mitochondrial matrix leads to loss of the membrane potential, swelling of the mitochondrial matrix, respiratory chain damage and a consequent decrease of oxidative phosphorylation, This results in diminished ATP production, free radical generation, and EMM rupture, followed by release of calcium and apoptotic factors [59,60]. These include molecules from the BCL-2 family (Bax, Bad, amongst others) [61], cytochrome C, pro-caspases 2, 3 and 9, apoptosis inducing factor and the second caspase activator derived from Smac/Diablo proteins [62]; which then activate caspase-dependent apoptosis or autophagy in the cytosol [63].
Delayed calcium dysregulation: This phenomenon describes the sudden and irreversible increase of cytosolic calcium levels that occurs after a period of stability in Glu concentrations. It is apparently caused by intramitochondrial calcium accumulation [64]. Although the mechanisms that lead to this phenomenon have not been clearly established, delayed calcium dysregulation appears to be the final step in a pathway that includes mitochondrial calcium overload and oxidative damage mediated by ROS [65]. Furthermore, it may act as a “point of no return” in neuron death, as it indicates that the calcium regulation has been damaged irreversibly, with release of cell death-promoting messengers from these organelles. Nonetheless, delayed calcium dysregulation does not appear to occur in all mitochondria within a neuron. It is estimated that approximately 35% of these organelles need to be affected before triggering irreversible cellular death [66].
Phospholipase activation: Activation of these and other enzymes is a consequence of calcium influx to neurons subjected to supraphysiologic Glu concentrations [67]. These enzymes are found in the cytosol, and contribute to neuronal membrane destruction through enzymatic lipid peroxidation [68]. Activation of phospholipase A generates arachidonic acid and its corresponding metabolites. These inhibit Glu uptake from the synaptic cleft, which leads to a continued activation of Glu receptors and a greater production of arachidonic acid. Increased levels of this molecule also form free radicals, which then activate phospholipase A, constituting a positive feedback loop. This cycle leads to apoptosis and autophagy, as well as formation of free radicals and lipid peroxidation [69].
Protease activation: Calpains are a family of calcium-activated cysteine-proteases located in the cytosol and the neuron synaptic terminal [70]. One of their products are free radical, after converting xanthine dehydrogenase into xanthine oxidase [71], an important enzyme in superoxide and hydroxyl (OH-) radical production. Damage occurs when free radical concentrations surpass the processing capacity of the cellular antioxidant systems, which leads to oxidative stress and cellular death. Unlike other organs, the brain is especially sensitive to free radicals because it has less endogenous antioxidant systems [72]. The most common free radicals are superoxide, peroxynitrite and OH-. These imbalances result in neuronal death as they damage phospholipidic membranes [73]. Similarly, free radicals may also damage DNA, which may occur acutely in ischemia, or chronically as part of the evolution of neurodegenerative diseases. Oxidative damage to DNA consists in DNA fragmentation and modification of nitrogenous bases [74]. These injuries may be reversible, except for damage caused to RNA [75].
Nitric oxide synthase: NMDARs are coupled to neuronal nitric oxide synthase (nNOS), whose activation results in an increase of nitric oxide (NO) concentration [76,77]. Toxic NO levels mediated by its metabolite, peroxynitrite [78], promote DNA fragmentation and interruption of protein kinase signaling. Moreover, in aqueous solutions, peroxynitrite spontaneously converts to OH-, promoting lipid peroxidation [71].
Protein Kinase C (PKC): The decrease in ATP levels and increase in free radical production boosts activation of the various subtypes of PKC [79]. Activation of γPKC triggers its translocation to the neuronal membrane, where it enhances postsynaptic NMDAR Glu sensitivity. Specifically, it acts by phosphorylating its NR1 subunit, perpetuating Glu influx to neurons [80]. On the other hand, activation and translocation of δPKC to the membrane activates BAD –a member of the Bcl-2 family–, cytochrome C and free radicals release. Cytochrome C release promotes apoptosis by caspase activation [81]. Furthermore, damage to mitochondrial PKC results in the loss of ATP regeneration and greater production of free radicals, promoting cell death [82].
Death in Slow Motion: Mechanisms Specific to Chronic Excitotoxicity
EAAT dysfunction
Deletion of the gene that codifies EAAT2 in mice will cause a 5% decrease in Glu uptake and its subsequent extracellular accumulation, with severe consequences such as hyperactivity, seizures, and restricted growth and premature death when compared to EAAT2-expressing mice [83]. Although EAAT2 has not been reported in humans; EAAT2 mutations may participate in disorders such as schizophrenia, alcoholism, bipolar disorder and ALS [40,84].
EAAT2 regulation
EAAT2 activity is regulated by numerous transcriptional and non-transcriptional mechanisms [85]. Amongst the former, repressor mechanisms include Nuclear Factor Kappa B and TNF alpha signaling. It has been observed that astrocytes in isolated cultures have do not express EAAT2, whereas those in neuron and astrocyte cultures show decreased EAAT2 expression after neuronal destruction. This highlights the necessary presence of neurons and glia cells for the expression of these transporters [86]. Epigenetic factors are also involved in EAAT2 expression, including CpG promoter methylation, which is lower in astrocytes in isolated cultures as well [87].
Non-transcriptional mechanisms include relocalization of transporters in response to signaling molecules, mainly PKC, that will act in positive and negative membrane EAATs “cluster” sites [88-90]. Because EAATs only act when located in the plasmatic membrane, their redistribution to this site results in potentiated Glu uptake [91].
In AD models, ARNm levels of EAAT2 appear to be normal, suggesting EAAT2 dysfunction caused by a post-transcriptional regulation mechanism [92]. In a mice model, utilization of a translational activator for EAAT2 restored EAAT2 function, with decreased risk for premature death, memory loss and amyloid β peptide accumulation [93].
Excitotoxicity: Clinical Implications
Excitotoxicity plays a fundamental role in neurodegenerative chronic disorders (Table 1), where a decrease in energy levels is prominent [94].
| Author/year(Reference) |
Type of study and Characteristics |
Conclusión |
| Zhang H. et al. [101] |
Huntington Disease: Experimental study In whichCa2+ signal pathways were studied on primary striatal medium spiny neuron cultures derived from murinesthatexpressedthe full-length human Huntingtin protein and displayed age-dependent loss of striatal neurons; and murineswhichexpressed a mutant Huntingtin protein without behavioral abnormalities or striatalneurodegeneration. To analyze pathways, Fura-2 Ca2+ imaging experiments were developed. |
AfterthefirstGlu pulse, supranormal Ca2+ responses were observed in murine neurons with full-length human Huntingtin; they were sensitized to Glu-induced apoptosis, and it induced rapid loss of mitochondrial membrane potential. Therefore, disturbed neuronal Ca2+ signaling plays a significant role in the degeneration of striatal medium spiny neurons containing full length mutant Huntingtin. |
Helton T. et al
[44] |
Parkinson’sDisease: Experimental studyto determine the effects of postsynaptic parkin on synaptic transmission on cultured rat hippocampal neurons which express GFP-tagged parkin. A whole-cell voltage clamp was used to record miniature excitatory postsynaptic currents and miniature inhibitory postsynaptic currents. |
Postsynaptic expression of parkin decreased excitatory synaptic transmission and caused a pronounced loss of excitatory synapses. Conversely, knockdown of endogenous parkin mutants deeply improved synaptic efficacy and precipitate proliferation of glutamatergic synapses, which increased vulnerability to synaptic excitotoxicity. |
Mitani y Tanaka
[37] |
AcuteIschemia: Experimental studydevelopedonmurineswith a glialglutamatetransporter (GLT-1) mutation, and wild-typemurinesto determine the function of GLT-1 during brain ischemia using an in vivo brain microdialysis technique. |
Glu levels in mice lacking GLT-1 were significantly higher than wild-type mice during 5 min ischemia. Excitotoxicity was induced on mutant murine but not in the normal one. When ischemia was extended to 20 min, Glu levels in wild-type mice were significantly higher than mutant. Acute neuronal death was also observed in the former. These suggest that GLT-1 has a protective function in the early stages of ischemia, however, when this is elongated, Glu is released and it precipitates acute neuronal death. |
D’Orsi et al.
[102] |
AcuteIschemia: Experimental studywhere the role of calpain activation during NMDA-induced excitotoxic injury was analyzed in embryonic murine cortical neurons that undergo excitotoxic necrosis, underwent excitotoxic apoptosis, or were tolerant to excitotoxic injury. Afterwards, they were treated with calpain inhibitors or calpain gene silencing, and monitorized through fluorescence microscopy using a calpain-sensitive Förster resonance energy transfer probe. |
Significant Calpain activity was not detected during excitotoxic necrosis or in neurons that were tolerant to excitotoxic injury. And they demonstrated that calpains were specifically activated during Bax-dependent apoptosis and that it has a role of downstream cell-death executioners. |
Table 1: Main excitotoxicity related diseases.
Alzheimer’s disease
In AD, Glu exerts tonic activation of NMDARs, which generates neuronal damage through calcium-dependent stimulation of catabolic enzymes [95]. Exaggerated hippocampus neuron activity could be an early sign of AD-related neuron degeneration [96]. At the molecular level, the hallmark β-amyloid plaques found in AD have been related to greater Glu synaptic release [97]. In addition, β-amyloid plaques also appear to activate or sensitize NMDARs, and conversely, NMDAR activation may promote β-amyloid synthesis [98]. Furthermore, continuous activation of NMDARs has been observed to impair long-term neuroplasticity which is required for memory formation [99].
These phenomena constitute the basis for the use of NMDAR antagonists-essentially memantine-in the treatment of AD, memantine is a non-competitive, voltage-dependent NMDAR antagonist [100]. The low-moderate affinity this molecule displays for NMDARs s particularly important for its beneficial role in AD, as it allows for NMDAR blocking preferentially when it is excessively open, and is fast-off the receptor, therefore inhibiting hyperactivation while retaining sufficient basal activation to prevent disruption of normal synaptic activity [101-103]. Indeed, memantine is regarded as a more tolerable alternative to other NMDAR antagonists, such as ketamine, phencyclidine and MK- 801, which do not match these pharmacodynamic properties [104]. Memantine has been observed to improve cognition, behavior, and daily functioning in subjects with AD, especially in moderate-severe cases [104,105]. Nevertheless, its effect size appears to be reduced [106], and it significantly increases the risk for side effects such as somnolence, weight gain, hypertension, falls and various nervous system disorders [107]. Thus, the search for further NMDAR antagonists with improved clinical utility or tolerability remains subject to intense ongoing research [108].
Amyotrophic lateral sclerosis
Excitotoxicity has been observed in subjects with motor neuron disease, with findings of Glu in the cerebrospinal fluid of individuals with ALS [109]. Similarly, beneficial effects for these patients have been found after the use of riluzole, an FDA approved drug that targets pathways related with ion transportation, including inhibition of Glu release at the presynaptic level [110]. Studies in rat hypoglossal motor neurons have suggested that increased levels of glycine might play a role in increased excitotoxicity in said neurons [111]. An antagonist for the glycine site on the NMDAR could therefore be used as a possible therapy for excitotoxicity reduction (REF). Functional defects in high-affinity sodium-dependent transporters responsible for Glu uptake have also been found in synaptosomes in the spinal cord and affected brain areas of ALS patients [112].
Parkinson’s disease
On the other hand, in Parkinson’s disease, excitotoxicity may be mediated by parkin, a protein codified by the PARK2 gene, which regulates stability and function of the excitatory Glu synapses. Postsynaptic parkin acts as a buffer for neuronal excitation. In contrast, parkin inexpression or mutant expressions are linked with increased synaptic efficiency and the proliferation of Glu synapses, which generates an elevated vulnerability towards excitotoxic processes in key sites, such as the substantia nigra [1]. Dopaminergic neurons in the CNS vulnerable to Parkinson’s disease exhibit bursting activity with increased activation of NMDARs expressing the NR1 subunit in higher than normal quantities [113].
Huntington’s disease
Finally, excitotoxicity has also been implicated in Huntington’s Disease (HD). Genetic studies in transgenic mice demonstrated that there is greater inclination towards neuronal death mediated by NMDARs in mice with HD, specifically through caspase 3-mediated apoptosis [114,115]. An increased number of postsynaptic NMDARs are present in the synapses, potentiating an excitotoxic effect. More specifically, the activation of the GluN2B subunit of NMDARs has been identified as a promoter of excitotoxicity induced by mutant huntingtin protein [116] which has been confirmed with the use of ifenprodil, a GluN2B antagonist in HD mouse cultured neurons in which it has been observed the absence of toxicity after administrating the drug [117].
Concluding Remarks
Analysis of neuronal death by excitotoxicity as a crime scene allows for the proposal of novel therapeutic approaches to diseases where this phenomenon is prominent. NMDA receptor antagonists are currently being explored in the context of an ample catalogue of disorders beyond AD, including epilepsy [118], depression [119] and hyperalgesia [120]. Recently, a role in NMDAR activation has been linked to depression and suicidal behavior, widening the consequences of the crime that is excitotoxicity [121,122]. Indeed, the future appears compelling for interventions based on NMDAR antagonism, which might be the way to redefine views on the management of a myriad of neuropsychiatric disorders.
19670
References
- Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: 379-87.
- Pivovarova NB, Andrews SB (2010) Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J 277: 3622-36.
- Lucas DR, Newhouse JP (1957) The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 58: 193-201.
- Olney JW (1969) Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164: 719-721.
- Watkins JC, Jane DE (2006) The glutamate story. Br J Pharmacol 147: S100-S108.
- van den Berg CJ, Garfinkel D (1971) A simulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J 123: 211-308.
- Won SJ, Kim DY, Gwag BJ (2002) Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol 35: 67-86.
- Auer RN (1986) Progress review: Hypoglycemic brain damage. Stroke 17: 699-708.
- Vincent P, Mulle C (2009) Kainate receptors in epilepsy and excitotoxicity. Neuroscience 158: 309-323.
- Fujikawa DG (2005) Prolonged seizures and cellular injury: understanding the connection. Epilepsy Behav 3: S3-S11.
- Ward RJ, Lallemand F, De Witte P (2009) Biochemical and neurotransmitter changes implicated in alcohol-induced brain damage in chronic or 'binge drinking' alcohol abuse. Alcohol Alcohol 44: 128-35.
- Cotter DR, Pariante CM, Everall IP (2001) Glial cell abnormalities in major psychiatric disorders: The evidence and implications. Brain Res Bull 55: 585-595.
- Olney JW (2003) Excitotoxicity, apoptosis and neuropsychiatric disorders.Curr Opin Pharmacol 3: 101-109.
- Leitch B (1992) Ultrastructure of electrical synapses: review. Electron Microsc Rev 5: 311-339.
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X (2010) Super-resolution imaging of chemical synapses in the brain. Neuron 68: 843-856.
- Blakely R, Edwards R (2012) Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harb Perspect Biol 4: a005595.
- Ramakrishnan NA, Drescher MJ, Drescher DG (2012) The SNARE complex in neuronal and sensory cells. Mol Cell Neurosci50: 58-69.
- Kew JN, Kemp JA (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Pyschopharmacology (Berl). 179 :4-29.
- Palmer LM, Shai AS, Reeve JE, Anderson HL, Paulsen O, et al. (2004) NMDA spikes enhance action potential generation during sensory input. Nat Neurosci 17: 383-90.
- Moriyama Y, Yamamoto A (2004) Glutamatergic chemical transmission: Look! here, there, and anywhere. J Biochem 135: 155-163.
- Tapiero H, Mathé G, Couvreur P, Tew K (2002) Glutamine and glutamate. Biomed Pharmacother 56: 446-457.
- Westergaard N, Sonnewald U, Schousboe A (1995) Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited. Dev Neurosci17: 203-211.
- Shachnai L, Shimamoto K, Kanner BI (2005) Sulfhydryl modification of cysteine mutants of a neuronal glutamate transporter reveals an inverse relationship between sodium dependent conformational changes and the glutamate-gated anion conductance. Neuropharmacology 49: 862-871.
- Shen J (2013) Modeling the glutamate-glutamine neurotransmitter cycle. Front Neuroenergetics 28: 5-10.
- Doyle KP, Simon RP, Stenzel-Poore MP (2008) Neuropharmacology – special Issue on cerebral ischemia mechanisms of ischemic brain damage – review article. Neuropharmacology 55: 310-318.
- Dave KR, Bhattacharya SK, Saul I, DeFazio R, Dezfulian C (2011) Activation of protein kinase c delta following cerebral ischemia leads to release of cytochrome c from the mitochondria via bad pathway. PLoS One 6: e22057.
- Phillis JW, Ren J, O’Regan MH (2000) Transporter reversal as a mechanism of glutamate release from the ischemic rat cerebral cortex: studies with DL-threo-beta-benzyloxyaspartate. Brain Res 868: 105-112.
- Shean RK, Lau CL, Shin YS, O’Shea RD, Beart PM (2013) Links between L-glutamate transporters, Na+/K+ATPase and cytoskeleton in astrocytes: evidence followinginhibition with rottlerin. Neuroscience 254: 335-346.
- Dingledine R, Borges K, Bowie D, Traynelis S (1999) The Glutamate Receptor Ion Channels. Pharmacol Rev 5: 7-62.
- Danysz W, Parsons CG (1998) Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev 50: 597-664.
- Tang CM, Dichter M, Morad M (1990) Modulation of the N-methyl-D-aspartate channel by extracellular H+. Proc Natl Acad Sci USA 87: 6445-6449.
- Williams K, Romano C, Dichter M, Molinoff PB (1991)Modulation of the NMDA receptor by polyamines. Life Sci 48: 469-498.
- Hume R, Dingldine R, Heinemann SF (1991)Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 253: 1028-1031.
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50: 295-322.
- Abdul-Ghani M, Valiante T, Carlen P, Pennefather P (1996)Metabotropic glutamate receptors coupled to IP3 production mediate inhibition of IAHP in rat dentate granule neurons. J Neurophysiol 76: 2691-2700.
- Mitani A, Tanaka K (2003) Functional changes of glial glutamate transporter GLT-1 during ischemia: An in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci 23: 7176–7182.
- Takahashi K, Foster J, Lin C (2015) Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci 72: 3489-3506.
- Beart P, O’Shea R (2007) Transporters for L-glutamate: An update on their molecular pharmacology and pathological involvement. Br JPharmacol150: 5–17.
- Zhou Y, Danbolt N (2013) GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 4: 165.
- Foran E, Trotti D (2009) Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid Redox Signal 11: 1587–1602.
- Veruki M, Morkve S, Hartveit E (2006) Activation of a presynaptic glutamatetransporter regulates synaptic transmission through electrical signaling. Nat Neurosci 9: 1388–1396.
- Kanai Y, Hediger M (2003) The glutamate and neutral amino acid transporter family: physiological and pharmacological implications. Eur J Pharmacol 479: 237-247.
- Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD (2008) Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci U S A 5: 19492-19497.
- Berridge M, Bootman M, Roderick H (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517-529.
- Bronner F (2001) Extracellular and Intracellular Regulation of Calcium Homeostasis. ScientificWorldJournal 1: 919-925.
- Mark L, Prost R, Ulmer J, Smith M, Daniels DL (2001) Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am J Neuroradiol 22: 1813-1824.
- Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey NW (2010) Modes of Neuronal Calcium Entry and Homeostasis following Cerebral Ischemia. Stroke Res Treat 316862.
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, et al. (2009) Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci USA 106: 9854-9859.
- Puyal J, Ginet V, Clarke PG (2013) Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 105: 24-48.
- Novelli A, Reilly J, Lysko P, HenneberryR (1988) Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res 451: 205-212.
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4: 552-565.
- Leung A, Halestrap A (2008) Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta 1777: 946-952.
- Leung A, Varanyuwatana P, Halestrap A (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol. Chem 283: 26312-26323.
- Crompton M (19993) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341: 233-249.
- Duchen M (2004) Roles of mitochondria in health and disease. Diabetes 53: 96-102.
- Szabo I, De Pinto V, Zoratti M (1993) The mitochondrial permeability transition pore may comprise VDAC molecules: II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 330: 206-210.
- Du H, Yan S (2010) Mitochondrial permeability transition pore in Alzheimer's disease: Cyclophilin D and amyloid beta. Biochim Biophys Acta.1802: 198-204.
- Koteswara V, Carlson E, Shidu S (2014) Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta 1842: 1267-1272.
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, et al.(2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 273: 2077-99.
- Gross A, McDonnell J, Korsmeyer S (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13: 1899-1911.
- Guo Y, Srinivasula S, Druilhe A, Fernades-alnemri T, Alnemri E (2002) Caspase-2 Induces Apoptosis by Releasing Proapoptotic Proteins from Mitochondria. J Biol Chem 277: 13430-13437
- Kim I, Rodriguez-Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245-253.
- Nicholls D, Budd S (1998) Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta 97-112.
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315-360.
- Pivovarova N, Nguyen H, Winters C, Brantner C, Smith C, et al. (2004) Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci 24: 5611–5622.
- Ong WY, Tanaka K, Dawe GS, Ittner LM, Farooqui AA (2013) Slow excitotoxicity in Alzheimer's disease. J Alzheimers Dis 35: 643-668.
- Hossmann KA (1996) Periinfarct Depolarizations. Cerebrosvasc brain Metab Rev 8: 195-208.
- Lipton S, Yeh M, Dreyer E (1994) Update on current models of HIV-related neuronal injury: platelet-activating factor, arachidonic acid and nitric oxide. Adv Neuroimmunol 4: 181–188.
- Melloni, Edon, Pontremoli, Sandro (1989) The calpains. Trends Neurosci 12: 438-444.
- Gagliardi RJ (2000) Neuroprotection, excitotoxicicity and nmda antagonists. Arq. Neuro-Psiquiatr 58: 583-588.
- Coyle J, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689–695.
- Farooqui T, Farooqui AA (2009) Aging: an important factor for the pathogenesis of neurodegenerative diseases. Mech Ageing Dev 130: 203-215.
- Cui J, Holmes E, Greene T, Liu P (2000) Oxidative DNA damage precedes DNA fragmentation afterexperimental stroke in rat brain. FASEB J 14: 955-967.
- Cui J, Holmes E, Liu P (1999) Oxidative damage to the c-fos gene and reduction of its transcription after focal cerebral ischemia. J Neurochem 73: 1164-1174.
- Brennan-Minnella AM, Shen Y, El-Benna J, Swanson RA (2013) Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis 4: e580.
- Breton R, Garcia J (2012) Excitotoxicity and Oxidative Stress in Acute Ischemic Stroke. Acute Ischemic Stroke 29-59.
- Dong X, Wang Y, Qin Z (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: 379-387.
- Zhao H, Ren C, Chen X, Shen J (2012) From rapid to delayed and remote postconditioning: the evolving concept of ischemic postconditioning in brain ischemia. Curr Drug Targets 13: 173-187.
- Bruno V, Battaglia G, Coupon A, Cespedes VM, Galindo MF, et al.(2001) An activity-dependent switch from facilitation to inhibition in the control of excitotoxicity by group I metabotropic glutamate receptors. Eur J Neurosci 13: 1469–1478.
- Dave K, Bhattacharya S, Saul I, DeFazio A, Dezfulian C (2011) Activation of Protein Kinase C Delta following Cerebral Ischemia Leads to Release of Cytochrome C from the Mitochondria via Bad Pathway. PLoS One 6: e2205.
- Bright R, Mochly-Rosen D (2005) The Role of Protein Kinase C in Cerebral Ischemic and Reperfusion Injury.Stroke 36: 2781-2790.
- Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, et al.(2006) From the cover: indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci USA 103: 12161-12166.
- Trotti D, Rolfs A, Danbolt NC, Brown RH Jr, Hediger MA (1999) SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci 2: 427-433.
- Kim K, Lee SG, Kegelman T, Su ZZ, Das S, et al. (2011) Role of excitatory amino acid transporter-2 (eaat2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol 226: 2484-2493.
- Takahashi K, Foster JB, Lin CL (2015) Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci 72: 3489-3506.
- Karki P, Smith K, Johnson J, Aschner M, Lee E (2015) Genetic dys-regulation of astrocytic glutamate transporter EAAT2 and its implications in neurological disorders and manganese toxicity. Neurochem Res 40: 380-388.
- Nakagawa T, Otsubo Y, Yatani Y, Shirakawa H, Kaneko S. et al.(2008) Mechanisms of substrate transport-induced clustering of a glial glutamate transporter GLT-1 in astroglial-neuronal cultures. Eur J Neurosci 28: 1719-1730.
- Zschocke J, Allritz C, Engele J, Rein T (2007) DNA methylation dependent silencing of the human glutamate transporter EAAT2 gene in glial cells. Glia 55: 663-674.
- Yang Y, Gozen O, Vidensky S (2010) Epigenetic Regulation of Neuron-Dependent Induction of Astroglial Synaptic Protein GLT1. Glia. 58: 277-286.
- Sheldon A, Robinson M (2007) The Role of Glutamate Transporters in Neurodegenerative Diseases and Potential Opportunities for Intervention. Neurochem Int 51: 333–355.
- von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J (2015) Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 7: 124.
- Takahashi K (2015) Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J Exp Med 212: 319-332.
- Beal MF (1992) Mechanisms of excitotoxicity in neurologic diseases. FASEB J 6: 3338-3344.
- Parsons CG, Stöffler A, Danysz W (2007) Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology 53: 699-723.
- Putcha D, Brickhouse M, O'Keefe K, Sullivan C, Rentz D, et al. (2011) Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non-demented elderly adults. J Neurosci 31: 17680-17688.
- Farfara D, Lifshitz V, Frenkel D (2003) Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease. J Cell Mol Med 12: 762-780.
- Arias C, Arrieta I, Tapia R (1995) Beta amyloid peptide fragment potentiates the calcium dependent release of excitatory amino acids from depolarized hippocampal slices. J Neurosci Res 41: 561-566.
- Newcomer JW, Farber NB, Olney JW (2000) NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci 2: 219-232.
- Robinson DM, Keating GM (2006) Memantine: a review of its use in Alzheimer's disease. Drugs 66: 1515-1534.
- Zhang H, Li Q, Graham R, Slow E, Hayden M (2008) Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington’s disease. Neurobiol Dis 31: 80-88.
- D'Orsi B, Bonner H, Tuffy LP, Düssmann H, Woods I, et al.(2012) Calpains are downstream effectors of baxdependent excitotoxic apoptosis. J Neurosci 32: 1847-1858
- Lipton SA (2005) The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res 2: 155-165.
- Rogawski MA, Wenk GL (2003) The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev 9: 275-308.
- Matsunaga S, Kishi T, Iwata N (2015) Memantine monotherapy for Alzheimer's disease: a systematic review and meta-analysis. PLoS One 10: e0123289.
- Areosa SA, Sherriff F (2003) Memantine for dementia. Cochrane Database Syst Rev 3: CD003154.
- Yang Z, Zhou X, Zhang Q (2013) Effectiveness and safety of memantine treatment for Alzheimer's disease. J Alzheimers Dis 36: 445-458.
- Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, et al.(2012) Novel NMDA receptor modulators: an update. Expert Opin Ther Pat 22: 1337-1352.
- Wuolikainen A, Moritz T, Marklund S, Antti H, Andersen P (2011) Disease related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE 6: e17947.
- Cheah B, Vucic S, Krishnan A, Kiernan M. Riluzole (2010) Neuroprotection and amyotrophic lateral sclerosis. Curr Med. Chem 17: 1942-1199.
- Paul P, de Belleroche J (2914) The role of D-serine and glycine as co-agonists of NMDA receptors in motor neuron degeneration and amyotrophic lateral sclerosis (ALS). Front Synaptic Neurosci 4: 6: 10.
- Tefera TW, Borges K (2016) Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front in Neurosci 10: 611.
- Roselli F, Caroni P (2015) From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85: 901-910.
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, et al. (2002) Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33: 849-860.
- Zhang H, Li Q, Graham RK, Slow E, Hayden MR, et al. (2008) Full length mutant huntingtin is required for altered Ca2+ signaling and apoptosis of striatal neurons in the YAC mouse model of Huntington's disease. Neurobiol Dis 31: 80-88.
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 14: 383-400.
- Sepers M, Raymond L (2014) Mechanisms of synaptic dysfunction and excitotoxicity in Huntington's disease. Drug Discovery Today 19: 990-996.
- Bethesda (MD) (2000) National Library of Medicine (US). ClinicalTrials.gov.
- Bethesda (2009) Does memantine improve verbal memory task performance in subjects with partial epilepsy and memory dysfunction? Clinical Trials gov
- Bethesda (2011) Treatment resistant geriatric depression in primary care. Clinical Trials gov
- Bethesda (2014) Hyperalgesia and NMDA Receptor Antagonist. ClinicalTrials.gov.
- Brundin L, Sellgren C, Lim C, Grit J, Palsson E, et al. (2016) An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Translational Psychiatry 6: e865.

