Leandro Bueno Bergantin* and Afonso Caricati-Neto
Laboratory of Autonomic and Cardiovascular Pharmacology, Department of Pharmacology, Paulista School of Medicine, Federal University of São Paulo, São Paulo, Brazil
Corresponding Author:
Leandro Bueno Bergantin
Laboratory of Autonomic and Cardiovascular Pharmacology, Department of Pharmacology, Paulista School of Medicine, Federal University of São Paulo (UNIFESP), Rua Pedro de Toledo, 669-Vila Clementino, São Paulo, Brazil
Tel: 551155764973
E-mail: leanbio39@yahoo.com.br
Received date: 20 February 2017; Accepted date: 27 February 2017; Published date: 03 March 2017
Citation: Bergantin LB, Caricati-Neto A. From a “Eureka Insight” to Novel Concepts in Pharmaceutical Sciences: Role of Ca2+/cAMP Intracellular Signalling Interaction. Ann Clin Lab Res. 2017, 5:1.
It has been almost 4 years since we revealed the explanation for the enigma of the so-called “calcium paradox”. Our discovery of the involvement of Ca2+/cAMP signaling interaction in the regulation of neurotransmitter release, and neuroprotection, was clearly a serendipitous discovery. It has produced new avenues in the understanding of the cellular and molecular mechanisms involved in the pathogenesis of neurological and psychiatric disorders, such as Alzheimer´s disease. Interestingly, this discovery initiated decades ago when numerous clinical studies have reported that use of L-type Ca2+ channel blockers (CCBs) by hypertensive patients decreased arterial pressure, but produced typical symptoms of sympathetic hyperactivity, such as tachycardia and increment of catecholamine plasma levels. Despite these adverse effects of CCBs have been initially attributed to adjust reflex of arterial pressure, during almost four decades this enigmatic phenomenon (the so-called "calcium paradox") remained unclear. In 2013, through an ingenious experiment, we discovered that this phenomenon was resulting of increment of transmitter release from sympathetic neurons, and adrenal chromaffin cells, stimulated by CCBs due to its interference on the Ca2+/cAMP signaling interaction. In this way, our discovery of the role of Ca2+/cAMP intracellular signaling interaction in the neurotransmitter release, and neuronal death triggered by cytosolic Ca2+ overload, opened novel adventures for the development of new pharmacological strategies more effective for the treatment of neurological and psychiatric disorders resulting of neurotransmitter release deficit, and neuronal death. These novel concepts have been extensively documented in several cited international papers of our own authorship (Bergantin and Caricati-Neto), and in an international book.
Keywords
Ca2+/cAMP signaling interaction; Calcium paradox; Neurological/psychiatric disorders
Introduction
The notion of stimulus-secretion coupling to explain neurotransmitters and hormones release has been resulted from ingenious experiments performed by Douglas and Rubin [1]. Complementing their concepts, Baker and Knight revealed in 1970´s that a rise in the cytosolic Ca2+ concentration ([Ca2+]c) is an elementary requirement to trigger transmitter release [2]. Indeed, the definite demonstration of a direct relationship between neurotransmitter release and increase in [Ca2+]c derived from the fundamental experiments performed by the Nobel laureate Erwin Neher [3]. More recently, many results have shown that cAMP increases neurotransmitter release at many synapses in autonomic nervous system of vertebrate, including sympathetic neurons [4]. Although the cellular mechanisms involved in these enhancer effects of cAMP on the release of neurotransmitters and hormones are under debate, the evidences indicate that this important intracellular messenger modulates signalling pathways mediated by Ca2+ involved in the regulation of neurotransmitter, and hormones release.
The Ca2+/cAMP signalling interaction as a universally-operated concept
The interaction between the intracellular signalling pathways mediated by Ca2+ and cAMP, named Ca2+/cAMP signalling interaction, has been widely studied in different cell types and tissues. This now a days accepted concept assumes that this interaction results in synergistic actions of these intracellular messengers on cell functions regulated by adenylyl cyclases (ACs), and phosphodiesterases (PDEs) [5-8]. The Ca2+/cAMP signalling interaction has particularly been extensively studied at the endoplasmic reticulum (ER) Ca2+ channels, such as Ca2+ channels regulated by ryanodine receptors (RyR) [5-8]. Our own experiments established that Ca2+/cAMP signalling interaction plays a key role in the regulation of neurotransmitter release from neurons and neuroendocrine cells [5-8]. Then, dysfunctions of cellular homeostasis of Ca2+ and/or cAMP in these cells could result in the dysregulation of Ca2+/cAMP signalling interaction, and could be a novel therapeutic goal for medicines.
From a “eureka insight” to novel concepts in pharmaceutical sciences
Indeed, several medical studies have been evidencing that acute and chronic use of L-type Ca2+ channel blockers (CCBs) in the antihypertensive therapy, such as nifedipine and verapamil, decreased peripheral vascular resistance and arterial pressure arterial, but produced typical symptoms of sympathetic hyperactivity such as tachycardia, and increment of catecholamine plasma levels [9]. Despite these adverse effects of CCBs have been initially attributed to adjust reflex of arterial pressure, during almost four decades the cellular and molecular mechanisms involved this enigmatic phenomenon named "calcium paradox" remained without additional explanation.
In 2013, through an ingenious experiment, we discovered that the "calcium paradox" phenomenon was resulting of increment of transmitter release from sympathetic neurons, and adrenal chromaffin cells, stimulated by CCBs due to its interference on the Ca2+/cAMP signalling interaction [6]. Using isolated tissues richly innervated by sympathetic nerves (rat vas deferens) to exclude the influence of adjusting reflex, we showed that neurogenic responses of the vas deferens were completely inhibited by L-type CCBs in high concentrations (>1 μmol/L), but unpredictably, and paradoxically, potentiated in concentrations below 1 μmol/L, characterized by sympathetic hyperactivity induced by CCBs [10-12]. Our studies showed that this paradoxical sympathetic hyperactivity is caused by increment of neurotransmitter release from sympathetic neurons produced by L-type CCBs due to its interference on the Ca2+/cAMP signalling interaction [5-8] (Figure 1).
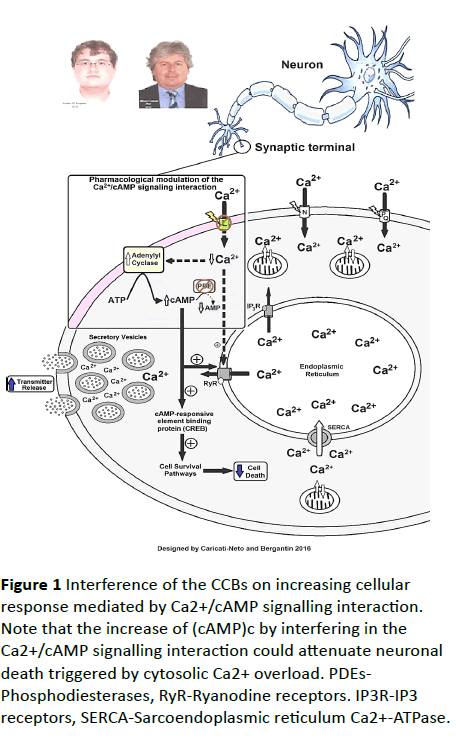
Figure 1 Interference of the CCBs on increasing cellular response mediated by Ca2+/cAMP signalling interaction. Note that the increase of (cAMP)c by interfering in the Ca2+/cAMP signalling interaction could attenuate neuronal death triggered by cytosolic Ca2+ overload. PDEs- Phosphodiesterases, RyR-Ryanodine receptors. IP3R-IP3 receptors, SERCA-Sarcoendoplasmic reticulum Ca2+-ATPase.
In addition, several studies have showed that increase of cytosolic cAMP concentration ([cAMP]c) stimulates neuroprotective response [13,14]. In this way, increase of (cAMP)c by interfering in the Ca2+/cAMP signalling interaction could attenuate neuronal death triggered by cytosolic Ca2+ overload [5-8]. Then, the pharmacological handling of the Ca2+/cAMP signalling interaction produced by combination of the L-type CCBs used in the antihypertensive therapy, and [cAMP]c enhancer compounds used in the anti-depressive therapy such as rolipram, could be a new pharmacological strategy for enhancing neurotransmission in neurological and psychiatric disorders resulting of neurotransmitter release deficit, and/or neuronal death [5-8]. These findings could open a new avenue for the drug development more effective and safer for the treatment of Alzheimer´s diseases [15-21].
Conclusion
From a “eureka insight” to novel concepts in pharmaceutical sciences. Pharmacological handling of the Ca2+/cAMP signalling interaction could be a more efficient and safer therapeutic strategy for stimulating neurotransmission compromised by neurotransmitter release deficit, and attenuating neuronal death.
Disclosure Statement
Caricati-Neto and Bergantin thank the continued financial support from CAPES, CNPq and FAPESP (Bergantin´s Postdoctoral Fellowship FAPESP #2014/10274-3).
The authors also thank Elsevier - “author use”:
Reuse of portions or extracts from the article in other works- https://www.elsevier.com/__data/assets/pdf_file/ 0007/55654/AuthorUserRights.pdf.
18495
References
- Coffinier C, Gresh L, Fiette L (2002) System morphogenesis defects and liver dysfunction upon targeted deletion of HNF1β. Development 129: 1829-1883.
- Sparks E, Huppert K, Brown M (2010) Notch signaling regulate formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 51: 1391-1400.
- Raynaud P, Tate J, Callens C (2011) A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology 53: 1959-1966.
- Desmet VJ (1992) Congenital diseases of intrahepatic bile ducts: Variations on the theme "ductal plate malformation". Hepatology 16: 1069-1083.
- Kerr DN, Harrison CV, Sherlock S (1961) Congenital hepatic fibrosis. Quart jmed 30: 91-117.
- Dhameja N, Rai V, Singh R (2016) Congenital hepatic fibrosis : Report on two cases and its clinicopathological correlation.AnnPatholLab Med 3: 90-94.
- Veigel MC, Prescott-Focht J, Rodriguez MG (2009) Fibropolycystic liver disease in children. Pediatr Radiol 39: 317-327.
- Nakanuma Y (2012) Tutorial review for understanding of cholangiopathy. Int J Hepatol 9.
- Ozaki S, Sato Y, Yasoshima M (2005) Diffuse expression of heparin sulfate proteoglycan and connective tissue growth factor in fibrous septa with many mast cells relate to unresolving hepatic fibrosis of congenital hepatic fibrosis. Liver Int 25: 817-828.
- Gunay-Aygun M, Tuchman M, Font-Montgomery E (2010) PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab 99: 160-173.
- Shenoy P1, Zaki SA, Shanbag P (2014) Caroli's syndrome with autosomal recessive polycystic kidney disease. Saudi J Kidney Dis Transpl 25: 840-843.
- Turkbey B, Ocak I, Daryanani K (2009) Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). PediatrRadiol 39: 100-111.
- Bayraktar Y (2011) Portal ductopathy: clinical importance and nomenclature. World J Gastroenterol 17: 1410-1415.
- Telega G, Cronin D, Avner E (2013) New approaches to the ARPKD patient with dual kidney-liver complications.Pediatr Transplant 17: 328-335.
- Srinath A, Shneider B (2012) Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease. J PediatrGastroenterolNutr 54: 580-587.
- Patel JN, Gupta S, Fauzdar M (2015) Congenital hepatic fibrosis associated with polycystic kidney disease.J Liver 4: 171.
- Bergmann C, Senderek J, Sedlacek B (2003) Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am SocNephrol 14: 76-89.
- Kim I, Fu Y, Hui H (2008) Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am SocNephrol 19: 455-468.
- Wen G (2011) Congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Clin Trans Sci 4: 460-465.
- Shorbagi A, Bayraktar Y (2010) Experience of a single center with congenital hepatic fibrosis: A review of the literature. World J Gastroenterol 16: 683-690.
- Nazer D, Elzouki NH (2012) Congenital hepatic fibrosis. Textbook of clinical pediatrics. 2ndedn. Berlin-Heidelberg: Springer-Verlag 3: 2013-2016.
- Giouleme O, Nikolaidis N, Tziomalos K (2006) Ductal plate malformation and congenital hepatic fibrosis: Clinical and histological findings in four patients. Hepatol Res 2: 147-150.
- Jiang L, Fang P, Weemhoff J (2016) Evidence for a “Pathogenic Triumvirate” in congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Biomed Res Int.
- Sato Y, Ren XS, Nakanuma Y (2012) Caroli's disease: Current knowledge of its biliary pathogenesis obtained from an orthologous rat model. Int J Hepatol.
- Parkash A, Cheema H, Malik H (2016) Congenital hepatic fibrosis: Clinical presentation, laboratory features and management at a tertiary care hospital of Lahore JPMA 66: 984.
- Geramizadeh B, Keramati P, Bahador A (2010) Congenital hepatic fibrosis and need for liver transplantation. Int J Organ Transplant Med 1: 98-100.