Keywords
Gene polymorphisms, Autonomic, Neurocardiovascular, Sympathetic, Parasympathetic, Therapy
Introduction
The adaptation of an organism to changes in external and internal environments is regulated by the autonomic nervous system [1-4]. The neural control of cardiovascular (CV) system plays a major role in such adaptations, even if different humoral mechanisms also participate in this control. The fact that “cardiovascular diseases are the leading cause of death in the world today and will remain so by the year 2020” strongly supports the need for new insights into CV neural regulatory mechanisms [5]. Mechanisms of neural CV regulation can be studied on systemic level by using different techniques of monitoring CV variables [6-12]. On the other hand, a rapid advance is taking place in the field of the molecular genetics of autonomic disorders of CV system [6]. An integrative approach of these two fields promises new aspects on pathophysiological mechanisms of autonomic disorders of CV system.
CV autonomic disorders can be classified as specific syndromes, sympathetic versus vagally mediated disorders [13]. We adopted this classification for its simplicity and orientation towards causality of the disease.
Sympathetic disorders can be classified as
a.Diseases in which activation of catecholamine system worsens an independent pathological state (coronary artery disease, arrhythmias (long QT syndrome, sudden death) and heart failure), and
b.Diseases in which abnormal catecholaminergic function is etiologic (sympathetic neurocirculatory failure, hypertension, cardiac necrosis and cardiomiopathy.
Vagally mediated cardiovascular diseases are
a.Neurally mediated syncope (vasovagal syncope, carotid sinus hypersensitivity)
b.Vagally mediated atrial fibrillation Mechanisms of these diseases are complex and multisystemic, involving endocrine, metabolic and immune system having autonomic nervous system in a central place as the major coordinator of patophysiological process [14]. Autonomic nervous system, being an interface between external environment and internal milieu buffers external stressful influences and coordinates the above mentioned bodily systems in order to achieve new, adapted body equilibrium. This role is damaged in neurocardiovascular diseases and this damage is more clearly described on the systemic level [6] as the change in modulation and phase of sympathetic and parasympathetic branch. Not much is known about autonomic molecular mechanism of pathophysiological changes. This gap in knowledge is especially present in the light of the question why some patients are more and some less susceptible to these diseases. Neurotransmission is molecular process strongly dependent on genetic background which profiles the phenotype of autonomic nervous system, both it’s strong, protective and weak points. We applied the systematic candidate gene analysis of the molecular network participating in the neurotransmission of sympathetic and parasympathetic nervous system, while more detailed facts about these pathophysiological entities can be found in Golstein 2001 and Mathias & Bannister 2013.
This review focuses on the understanding of CV neurogenetics, in specific the genetic polymorphisms of the peripheral components of autonomic nervous system- sympathetic and parasympathetic signaling, metabolic pathways and molecular interactions. Their influence on regulatory mechanisms in autonomic disorders of CV system is of primary importance.
Sympathetically mediated cardiovascular disorders
1a. Candidate genes in sympathetic disorders in which activation of catecholamine system worsens an independent pathological state: Candidate genes for sympathetic disorders can be found all along the sympathetic signaling pathway (Figure 1) whose milestones are genes of adrenergic receptors (alpha1A (ADRA1A), alpha1B (ADRA1B), alpha1D (ADRA1D), alpha2A (ADRA2A), alpha2B (ADRA2B), alpha2C (ADRA2C), beta1 (ADRB1), beta2 (ADRB2), beta3 (ADRB3), dopamine receptor type 1A (DRD1A)), norepinephrine transporter (NET), G-proteins (Gs protein alpha subunit (GNAS1), G protein beta 3 subunit (GNB3). Other molecules, also a part of intracellular signaling pathways, like G protein related kinases (GRK 1-7) are interesting candidates for genetic evaluation, but are still on the very beginning of full elucidation of their role in neurocardiovascular diseases [6].
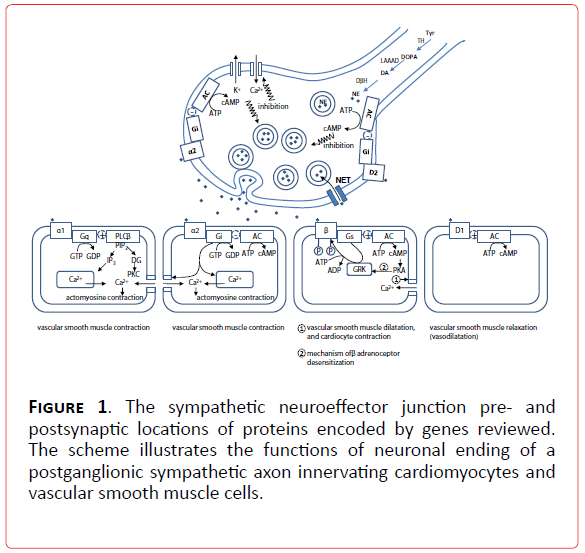
Figure 1. The sympathetic neuroeffector junction pre- and postsynaptic locations of proteins encoded by genes reviewed. The scheme illustrates the functions of neuronal ending of a postganglionic sympathetic axon innervating cardiomyocytes and vascular smooth muscle cells.
Another functionally coupled network of molecules is responsible for the complex metabolic dynamics of catecholamines involving: synthesis (tyrosine hydroxylase (TH), enzymes of tetrahydrobiopterine metabolism (GTP cyclohydrolase GCH1, dihydrobiopteridine reductase DHPR), L-aromatic-amino-acid decarboxylase (LAAAD), dopamine-beta-hydroxilase (DBH), copper ATPase), inactivation (monoamine oxydase (MAO) , catechol-O-methyltransferase (COMT), phenylethanolamine N-methyltransferase (PNMT), aldehyde dehydrogenase (ALDH1A1 and ALDH2)) and storage (vesicular monoamine transporter 2 (VMT2), H+vesicular ATPase and chromograninA-catestatin (CgA) [13-15] (Figure 2). These molecules deserve special attention for candidate gene studies( Table 1).
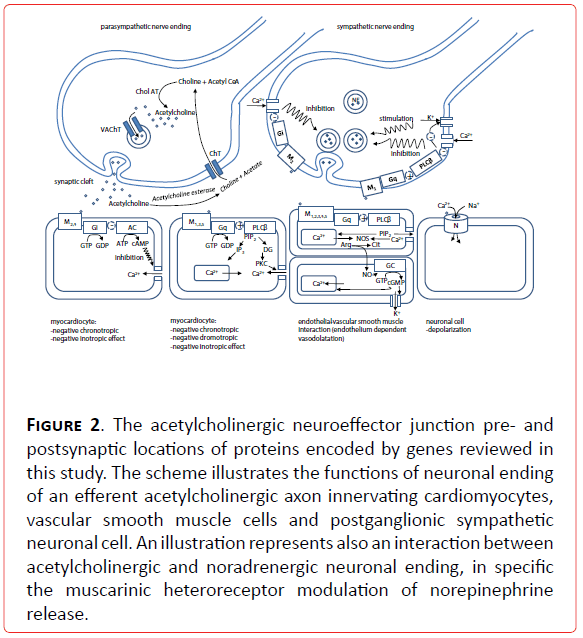
Figure 2. The acetylcholinergic neuroeffector junction pre- and postsynaptic locations of proteins encoded by genes reviewed in this study. The scheme illustrates the functions of neuronal ending of an efferent acetylcholinergic axon innervating cardiomyocytes, vascular smooth muscle cells and postganglionic sympathetic neuronal cell. An illustration represents also an interaction between acetylcholinergic and noradrenergic neuronal ending, in specific the muscarinic heteroreceptor modulation of norepinephrine release.
| Genes important for catecholaminergic neurotransmission |
Diseases in which activation of catecholamine system worsens an independent pathological state |
| CAD |
arrhythmias |
HF |
| ADRA1A |
|
|
|
| ADRA1B |
|
|
|
| ADRA1D |
|
|
|
| ADRA2A |
|
|
|
| ADRA2B |
D/D (Snapir et al., 2001)
D/D(Snapir et al., 2003) |
|
|
| ADRA2C |
Del 322-325 (Park et al., 2006) |
|
Del 322-325# (Shin et al., 2007)
Del322-325# (Savva et al., 2009) |
| ADRB1 |
Ser49Ser (Hassan et al., 2008)
Gly389Arg#(Park et al., 2006)
Codons 49 and 389(Hassan et al., 2008) |
Gly 389 Arg, Ser 49 Gly (Ulucan et al., 2008)
Ser49Gly, Arg389Gly#(Paavonen et al., 2007)
Ser49Gly,Gly389Arg # (Kanki et al., 2002) |
Ser49Gly*(Borjesson et al., 2000)
Ser49Gly(Podlowski et al., 2000)
Arg389# (Tesson et al., 1999)
Arg389(Mialet Perez et al., 2003)
Gly49Gly (Pereira et al., 2012)
S49G, R389G#(Shin et al., 2007) |
| ADRB2 |
46A(Zak et al., 2008)
Gln27(Park et al., 2006)
Arg16Gly#(Park et al., 2006)
Glu27*(Heckbert et al., 2003)
Codons 16,27 and nucleotide 523# (Hassan et al., 2008) |
Arg16Gly, Gln27Glu,Thr16Ile (Ulucan et al., 2008)
Thr184Ile#(Kanki et al., 2002)
Gln27Gln (Sotoodehnia et al., 2006)
Gly16Arg#(Sotoodehnia et al., 2006) |
Thr164Ile (Brodde et al., 2001)
Glu27Glu, Arg16Arg (Pereira et al., 2012)
Arg16Gln27 (Shin et al., 2007) |
| ADRB3 |
|
|
|
| DRD1A |
|
|
|
| NET |
|
|
|
| GNAS1 |
|
|
|
| GNB3 |
C825T ,1429G>A, IVS5+41G>A, 657T>A, 814G>A# (Renner et al., 2007)
C825T* (Michalsen et al., 2006) |
C825T(Wieneke et al., 2006)
T825T*(Schreieck et al., 2004) |
|
| GRK |
|
|
GRK5 Leu41* (Cresci et al., 2009) |
| Genes important for catecholaminergic metabolism |
|
|
|
| TH |
|
|
|
| GCH1 |
C+243T(Zhang et al., 2007) |
|
|
| DHPR |
|
|
|
| LAAAD |
|
|
|
| DBH |
|
|
|
| Cu2+ATPase |
|
|
|
| PNMT |
|
|
|
| MAO |
rs2576178 A > G , rs2296545 C > G # (Fava et al., 2012)
rs2296545 C > G (Farzaneh-Far et al., 2010) |
|
rs2296545 C > G (Farzaneh-Far et al., 2010) |
| COMT |
LL (Happonen et al., 2006) |
|
|
| ALDH |
ALDH2rs671 (G>A) ((Han et al., 2013)
ALDH2rs671 (G>A) (Wang et al., 2013) |
|
|
| H+ vesicular ATPase |
|
|
|
| VMAT2 |
|
|
|
| CgA-catestatine |
|
|
|
* - protective effect
# - no association was found.
Table 1. The association between candidate genes and neurocardiovascular diseases where the activation of catecholamine system worsens an independent pathological state
Coronary artery disease
Coronary artery disease (CAD) is a multifactorial disorder which results from the interactions between the number of genetic and non-genetic factors [16,17]. It includes the group of pathophysiological conditions where coronary oxygen supply is decreased or blocked, generally due to atherosclerotic plaque. Sympathetic overactivity, an important component of these syndromes, can be boosted by environmental conditions (psychological stress [18, 19], smoking [20] which interact with the genetically determined response pattern. The research on the role of genetic polymorphisms of the molecules in the sympathetic signaling pathway was put on adrenergic receptors, identifying ADRA as the critical place of sympathetic cytophysiology in CAD. Gene polymorhisms of these receptors which are responsible for coronary vasoconstriction (Figure 1), are the risk factors for the genesis of CAD. Less net results are obtained for ADRBs, the important mediators of vascular dilatation (Figure 1). The divergent results could be interpreted to be the consequence of the studies on different populations, different syndromes and parameters of CAD, and different impact of gene-environment interaction. An interesting result is obtained by [21], reporting a protective role of C825T polymorphism of GNB3 subunit in CAD. The protective role of this polymorphism was present in the circumstance of the lifestyle change [21], becoming in this way an indicator for behavioural therapy, a useful marker of the patient prognostic outcome and a potential target for pharmacotherapy research.
GCH1 is the rate limiting enzyme in the synthesis of tetrahydrobiopterine (BH4), an important cofactor for TH activity and NO dependent mechanism of acetylcholine vasodilatation [17], (Figure 3) The decreased rate of BH4 bioavailability may cause the dysfunction of both components of ANS (vagal and catecholaminergic) and join to this pathophysiological context an increased superoxide production, a known risk factor for atherosclerosis. The fact that 3 od 13 SNPs were found in promoter region of GCH1 speaks in favor that different polymorphisms might affect alternative tissues (vascular tissue vs. kidney) [22]. GCH1 genotypes might be useful in defining the specific phamacogenomic therapy in susceptible patients. Another gene relevant for BH4 bioavailability, DHPR, shows an association between its activity and acetylcholine induced vasodilatation, but till now no studies were performed on the association of DHPR SNPs and CAD [23]. The SNP of renalase, enzyme with MAO activity, was recently shown to be associated with inducible cardiac ischemia [24]. The study conducted on the enzyme of the adrenergic metabolism COMT showed that a heavy coffee intake increased the risk of CAD in patients with LL genotype pointing out on the importance of this gene-environment interaction [25].
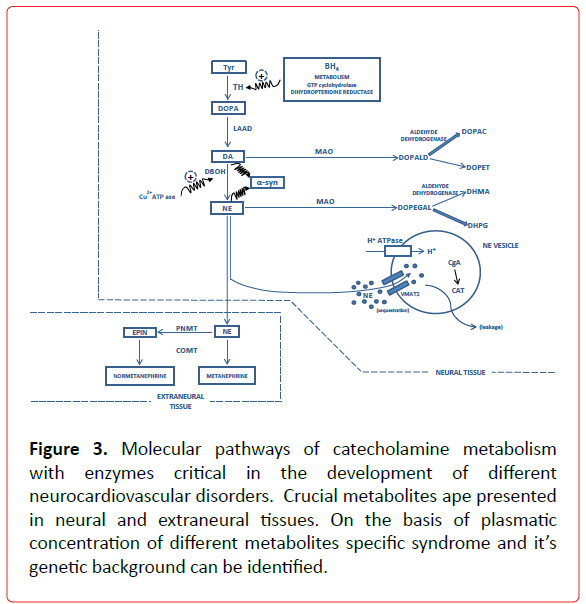
Figure 3. Molecular pathways of catecholamine metabolism with enzymes critical in the development of different neurocardiovascular disorders. Crucial metabolites ape presented in neural and extraneural tissues. On the basis of plasmatic concentration of different metabolites specific syndrome and it’s genetic background can be identified.
ALDH (ALD1A1 and ALDH2) are the important enzymes in the mechanism of the toxic aldehyde clearance in the circumstances of acute myocardial ischemia and have protective effect for reperfusion arrhythmogenesis [26]. A recent meta-analysis [27,28] provide convincing data about the association of ALDH2 SNP rs671 (G>A) with the risk of CAD and MI. Compartmentalization of monoamines into vesicles is the dominant mechanism of monoamine removal from cytoplasm and prevention of their toxic effects [15]. Dynamic interaction between different protein components of these compartments is essential for monoamine metabolism. The most studied proteins are VMAT2, H+ATPase and CgA-catestatin. VMAT2, H+ATPase and CgA-catestatin SNPs role in the CAD development still is missing.
Arrhythmias
Different types of arrhythmias were investigated for the role of SNPs as their risk factors. Due to their role in the modulation of signal transduction and arrhythmogenesis ADRB receptors deserved most of the attention. Idiopathic ventricular arrhythmias commonly refer to ventricular tachycardia and/or frequent/ monomorphic premature ventricular contractions in patients with structurally normal heart [29]. Activation of sympathetic tone has been shown to play an important role in the provocation and maintenance of these arrhythmias.
Negative result for the participation of ADRB as the risk factor in the genesis of torsades de pointes was obtained by [30]. An interesting study was performed by Paavonen et al., who investigated the association between ADRB SNPs with ECG characteristics of the patients with long QT syndrome and the risk of SCD [31]. No association was found and possible explanation of this result was that stimulation of ADRB potentiates the risk of SCD by different mechanism. Ulucan et al., found that patients with idiopathic ventricular arrhythmias had higher frequency of Arg389Arg genotype, Arg389Gly49, and Arg389Ser49 haplotypes of the ADRB1, and higher frequency of Gly16Gly, Glu27Glu genotypes and Gly16Gln27Thr164, Gly16Glu27Thr164, and Gly16Glu27Ile164 haplotypes of the ADRB2 compared to control subjects [32]. Their results show that common SNPs in the ADRB1 and ADRB2 are significantly associated with idiopathic ventricular arrhythmias in Turkish population.
One potential genetic marker for sudden cardiac arrest is located in a gene coding the G-protein. The G-protein participates in intracellular signaling cascades relevant also for the regulation of ventricular rhythm. Moreover, some evidence has indicated that the G-protein might be directly involved in the genesis of atrial and ventricular arrhythmias [33,34]. Wieneke et al., studied whether the C825T polymorphism in GNB3 might serve as a genetic marker for recurrent ventricular arrhythmias [32]. Homozygous carriers of the C825 allele had a 3.9-fold risk for severe ventricular arrhythmias. An interesting result was obtained by Schreiech et al., who investigated a possible impact of the GNB3 C825T polymorphism on atrial fibrillation in an association study [35]. They concluded that there was an association between the GNB3 TT genotype and a reduced risk for the occurrence of atrial fibrillation. The fact that GNB3 C825T polymorphism plays the opposing roles in the genesis of ventricular vs. atrial arrhythmias is in line with the fact that these arrhythmias have different pathophysiological mechanisms, with different roles of sympathetic and vagal influence on their genesis. G proteins are an important component of the signaling pathway of both sympathetic and parasympathetic nervous system and integrative G protein functioning might be crucial for the resulting cellular response on sympatho-vagal drives. Again, the different results on the role of ADRB and G protein polymorphisms are obtained in different experimental designs and on different populations. A detailed investigation on the role in arrhythmogenesis of above reported and other molecules of the sympathetic signaling pathway is necessary.
Although a significant association between the expression of GCH1 and heart rate was confirmed, no study was done about the association between different GCH1 SNPs and arrhythmias [36]. Considering the proteins of monoamine vesicular compartmentalization, an initial research is done on the role of VMAT2 in arrhythmogenesis [37]. Further studies are needed on the role of vesicular proteins SNPs in arrhythmias.
Heart failure
One of the main functions of the cardiac adrenergic system is to regulate cardiac work both in physiologic and pathologic states. A better understanding of this system has permitted the elucidation of its role in the development and progression of heart failure (HF). Enhanced sympathetic activation has a central role in the development and progression of HF [38].
Liggett et al., have demonstrated that adrenergic polymorphisms affect not only receptor signaling and sensitivity to pharmacological agents, but also influence clinical outcomes of HF; for ADRB1, two polymorphic loci have been investigated: Ser49Gly and Arg389Gly polymorphism [39,40]. Borjessen et al., also found that allelic distribution of Ser49Gly has been associated with long term survivor in patients with HF [41]. This is consistent with idea that downregulation and desensitization of ADRB1 receptors is protective in HF [42]. On the contrary, Podlowski et al., found that Ser49Gly is more frequent in patients with idiopathic dilated cardiomyopathy [43]. Another important ADRB1 polymorphism is a Arg389Gly polymorphism in the fourth intracellular loop, which participates in G protein coupling. The polymorphism Arg389 of Arg389Gly, is threefold more effective than is the Gly389 variant at activating adenylyl cyclase [44], which speak for the higher potential of Arg389 carriers in initiation and maintenance of cardiac hypertrophy. Still, no correlation between Arg389 and HF was observed by Tesson et al., [45], but more recent results of Mialet Perez et al., [46] show that when expressed Arg389, even at low level, in mouse hearts induces HF. More, the responses to antagonists are more pronounced in Arg389 than in Gly389. It is of interest that Arg389 significantly increases the cardiovascular risk of HF in subjects with ADRA2C deletion variant [47]. These data suggest the importance of interaction of individual polymorphisms in the development of pathological state.
ADRB2 are highly polymorphic. One rare variant, Thr164Ile is associated with reduced survival and depressed exercise capacity in HF patients [48]. In the study of Pereira et al., ADRB1 polymorphisms (Ser49Gly, Arg389Gly and Gln27Glu) and ADRB2 polymorphisms (Gln27Glu and Arg16Gly) were investigated for the association with HF progression, drug response and clinical outcome [49]. Genotypes Gly49Gly of the ADRB1 polymorphism and Glu27Glu and Arg16Arg of ADRB2 polymorphisms were higher in HF patients compared with healthy population. Their study showed the racial specificity where black population with Ser49Ser genotype showed a reduced survival rate with respect to other two genotypes of ADRB1.
The role of adrenergic receptors genetic polymorphisms in adverse events of the HF, defined as death or heart transplantation were investigated in the study of Shin et al., [50]. Eight polymorphisms in 6 genes were genotyped: ADRB1 (S49G, R389G), ADRB2 (G16R, Q27E), ADRA2C (insertion/deletion 322-325), angiotensinogen (AGT, M235T), angiotensin receptor type 1 (AGTR1, 1166A>C), and angiotensin-converting enzyme (ACE, insertion/deletion in intron 16). On the basis of this investigation, only ADRB2 Arg16Gln27 haplotype may significantly increase the risk of adverse outcomes in patients with HF receiving contemporary HF pharmacotherapy. Savva et al., investigated whether the ADRA2C (Del322-325) polymorphism exclusively or in combination with a ADRB1 (Arg389) polymorphism, each with known independent effects on sympathetic function, were associated with an increased risk of adverse events in HF [51]. The authors have found that the ADRA2C Del322-325 polymorphism exclusively or in combination with the ADRB1 Arg389 allele is not associated with an increased risk of adverse events in HF.
The contribution of adrenergic receptors to the development of HF needs to be more thoroughly investigated: studies designed to consider many complex haplotypes including the combination of individual polymorphisms are necessary.
Functionally significant gene variants of the G-protein receptor kinases (GRKs) that phosphorylate and uncouple ADRB receptors from their downstream signaling effectors were candidates for polymorphism studies. One common nonsynonymous polymorphism was found, substituting Leu for Gln at GRK5 amino acid 41 [52]. This polymorphism is rare in individuals of European descent, but approximately 40% of individuals of African ancestry are the carriers. This polymorphism increases ADRB1 and ADRB2 desensitization [52,53]. The first clinical study of the GRK5 Leu41 polymorphism showed that beta-blocker naive carriers had improved transplant-free survival [54]. The GRK5 interaction with HF has only been described in the absence of beta-blocker therapy, which is the standard of care for essentially all forms of HF [55].
Considering the association studies of SNPs important for the catecholamine metabolism and HF, the noticeable lack of data is present. The SNP of renalase, enzyme with MAO activity, was recently shown to be associated with clinical parameters of HF [24].
1b. Candidate genes in sympathetic disorders in which abnormal catecholaminergic function is etiologic (Table 2).
| Genes important for catecholaminergic neurotransmission |
Diseases in which abnormal catecholaminergic function is etiologic |
| SNF |
Hypertension |
Cardiac necrosis and cardiomyopathy |
| ADRA1A |
|
Cys/Cys #(Xie et al., 1999)
Cys/Cys (Freitas et al., 2008)
Cys/Cys *(Gu et al., 2006) |
|
| ADRA1B |
Gly 49*(Winker et al., 2005) |
|
|
| ADRA1D |
|
|
|
| ADRA2A |
|
|
|
| ADRA2B |
|
|
|
| ADRA2C |
|
|
Del322-325* (Regitz-Zagrosek et al., 2006)
Del 322-325# (Sharkey et al., 2009) |
| ADRB1 |
|
Ser49Gly# (Filigheddu et al., 2011)
Arg389Gly# (Filigheddu et al., 2011) |
Ser49Gly*, Arg389Gly* (Forleo et al., 2004)
rs35230616#, rs1801253# (Spinelli et al., 2010)
Ser49Gly#, Arg389Gly# (Sharkey et al., 2009)
Arg389Gly (Vriz et al., 2011)
rs1801253# (Figtree et al., 2013) |
| ADRB2 |
|
Arg16Gly (Kotanko et al., 1997)
Arg16Gly , Gln27Glu (Bray et al., 2000)
Arg16Gly (Busjahn et al., 2000)
Arg16Gly (Hoit et al., 2000)
Arg16Gly (Wu et al., 2006)
Arg16Gly#(Wu et al., 2006)
Cys19Arg# (Filigheddu et al., 2011)
Gly16Arg# (Filigheddu et al., 2011)
Gln27Glu# (Filigheddu et al., 2011) |
5' LC Arg19Cys*, Arg16Gly*, Gln27Glu*, and Thr164Ile# (Forleo et al., 2004)
rs1042713#, rs1042714#, and rs1800888# (Spinelli et al., 2010)
Gln27Glu (Vriz et al., 2011)
rs1800888# (Figtree et al., 2013) |
| ADRB3 |
|
|
Arg64Trp# (Forleo et al., 2004) |
| DRD1A |
|
|
|
| NET |
|
rs168924 (Ono et al., 2003)
rs168924# (Zolk et al., 2012) |
|
| GNAS1 |
|
Fokl(+/-) (Jia et al., 1999) |
rs11554276# (Spinelli et al., 2010) |
| GNB3 |
|
C825T (Beige et al., 1999)
C825T, C1429T(Cabadak et al., 2011)
G5177A# (Cabadak et al., 2011)
C825T# (Niu et al., 2011)
A3882C#, G5249A# and C825T# (Filigheddu et al., 2011) |
|
| GRK |
|
|
GRK5 rs17098707, rs34679178 (Spinelli et al., 2010)
GRK5 rs17098707# (Figtree et al., 2013) |
| Genes important for catecholaminergic metabolism |
|
|
|
| TH |
|
C-824T (Nielsen et al., 2010) |
|
| GCH1 |
|
C+243T (Zhang et al., 2007) |
|
| DHPR |
|
|
|
| LAAAD |
|
|
|
| DBH |
V87M, D100E, and D331N# (Cho et al., 2003) |
-1021C/T (Abe et al., 2005)
C-970T (Chen et al., 2010)
C-2073T (Chen et al., 2011) |
|
| Cu2+ATPase |
|
|
|
| PNMT |
|
PNMT-148, PNMT-353 (Cui et al., 2003)
G-367A, G-161A* (Huang et al., 2011) |
|
| MAO |
|
rs2296545 (Buraczynska et al., 2011)
rs2576178# (Buraczynska et al., 2011)
rs2576178, rs10887800 (Stec et al., 2012)
rs2576178 A > G, rs2296545 C > G# (Fava et al., 2012) |
|
| COMT |
|
-1187G>C(Kamide et al., 2007)
rs4680 (Hagen et al., 2007)
rs4680 (Annerbrink et al., 2008)
rs4680 (Htun et al., 2011) |
rs4680# (Figtree et al., 2013) |
| ALDH |
|
ALDH2 rs671# (Sen Zhang et al., 2013) |
|
| H+ vesicular ATPase |
|
|
|
| VMAT2 |
|
|
|
| CgA-catestatine |
|
|
|
* - protective effect
# - no association was found
Table 2. The association between candidate genes and neurocardiovascular diseases where abnormal catecholaminergic function is etiologic
Sympathetic neurocirculatory failure
Sympathetic neurocirculatory failure (SNF) features orthostatic hypotension and abnormal beat-to-beat blood pressure responses to the Valsalva maneuver. This pathophysiological phenomenon is present in Pure autonomic failure (or Bradbury- Eggleston syndrome or idiopathic orthostatic hypotension), Shy- Drager Syndrome (multiple system atrophy with sympathetic neurocirculatory failure) and Parkinsonism with peripheral autonomic failure [13].
Several features of idiopathic orthostatic hypotension have been attributed to hypoactive or hyperactive states of adrenergic receptors of the sympathetic nervous system. Focused on this fact, Winker et al., wanted to ascertain whether genotypes at eight polymorphic loci within five relevant adrenergic receptor genes (ADRA2A, ADRA2B, ADRA2C, ADRB1 and ADRB2) influence the risk for idiopathic orthostatic intolerance [56]. They found for the ADRB1 Gly49 polymorphism a decrease in the risk of idiopathic orthostatic intolerance among persons who were homozygous, implying its protective role. No other associations for other adrenergic receptors polymorphisms and idiopathic orthostatic intolerance were found.
The norepinephrine-synthesizing DBH is a reasonable candidate gene for autonomic diseases with sympathetic insufficiency [57]. On the basis of their previous findings, the authors hypothesized that mutations found on DBH locus might be risk factors for autonomic diseases like orthostatic intolerance, pure autonomic failure and multiple system atrophy. Three missense mutations (V87M, D100E, and D331N) were taken in consideration, but none of these mutations was revealed in reported autonomic syndromes. Even though the authors report the preliminary results, it is noteworthy that the frequency of the T allele mutation 1021C→T in orthostatic intolerance patients is slightly higher than that in controls. Some reasons for the lack of data on association studies between SNPs of catecholaminergic signaling and metabolic pathways and SNF, is due to the rarity of the syndrome and complexity of the clinical phenotype.
Hypertension
Essential hypertension is a multifactorial disease with increasing number of evidence that increased and adverse activation of sympathetic nervous disease plays a crucial role in it's genesis [14,38]. Hypertension candidate genes act through all the signaling and metabolic pathways of catecholaminergic function [58]. In an effort to localize blood pressure influencing genes, Krushkal et al., [59,60] identified the long arm of human chromosome 5 (5q33.3 to 34) as the region putatively containing ADRA1B, ADRB2 and DRD1A genes influencing systolic blood pressure levels in young Caucasians. Both ADRA1B and DRD1A genes are located close to these markers, suggesting that genetic variation in one or both of these receptors plays an important role in determining interindividual variation in systolic blood pressure levels. The same group [61] reported further data on association between the polymorphisms within the ADRB2 gene and systolic blood pressure among discordant siblings. Multiple lines of investigation indicate that the Arg16Gly and Gln27Glu polymorphisms in the ADRB2 gene significantly influence interindividual blood pressure variation and the occurrence of hypertension. Associations were detected between this polymorphisms and blood pressure in normotensive twins [62]. Studies by Hoit et al., add more evidence that the Gly16 variant imparts attenuated vasodilatory responses to catecholamines in humans, further emphasizing the ADRB2 gene role in the regulation of the arterial blood pressure [63]. There are also confirmed significant associations of essential hypertension in Yi minority group with separate SNPs of ADRB2 [64]. In contrast no association was found in the Hani minority group. These data confirmed that the mutations of this gene show ethnic, as well as population and even subpopulation specificity.
The association of ADRA1A polymorphisms with blood pressure phenotypes was studied in different populations: Brasilian [65], African American and Caucasian population [66]. These studies revealed controversial role of ADRA1A in association to essential hypertension: presence of Cys/Cys genotype was associated with hypertension only in individuals with regular physical activity, while an initial report of Xie et al., found no association between this polymorphism and hypertension [66]. Recent studies showed that the Cys allele was associated with relatively lower hypertension prevalence in a Chinese population [67]. Again, these results suggested that genetic variations in ADRA1A could modulate cardiac or vascular sympathetic modulation in highly population dependent manner.
The attempts have been made to investigate the implication of NET in hypertension pathophysiology. The proposed hypothesis was that reduced uptake of norepinephrine could provide an increased concentration of catecholamines in neuromuscular synaptic cleft and circulation that would initiate and maintain an increased vasoconstrictor response of vascular smooth muscle cells. Still, both experimental [68,69]) and clinical findings [70] about the role of NET in hypertension remain without straightforward conclusion. NET gene (SLC6A2) is located and mapped [71], and its active role in hypertension is confirmed (NET gene silencing, [72]. Numerous SNPs have been identified, but only rs168924 is associated with essential hypertension in Japanese population [73]. A detailed analysis of the interrelation of rs168924 polymorphisms of NET gene and blood pressure parameters in Caucasian population provided opposite resultsrs168924 G variant was associated with a lower prevalence of arterial hypertension [74]. There is also the possibility that some rare forms of hypertension, like hypernoradrenergic hypertension, could result from the genetic dysfunction of NET [75].
Several studies have reported the association of G protein polymorphisms and essential hypertension in some population, although contradictory results were reported. The study of Jia et al., demonstrates an association between a Fokl(+/-) polymorphism in the GNAS1 locus and hypertension [76]. More, the Fokl allele was more common in good responders than poor responders to blood pressure responses after beta-blocade. In the study of Cabadak et al., it was investigated the potential role of the C825T, C1429T, and G5177A polymorphisms of the GNB3 in essential hypertension in a group of Turkish subjects [77]. The frequencies of the 825T and 1429T variants were higher in hypertensive subjects compared to those of controls.
Metaanalysis of alpha-adducin G460T and GNB3 C825T polymorphisms in southern Chinese population showed null association with hypertension but indicate local marginal significance of C825T, as a putative salt-sensitive switch [78]. By analyzing polymorphisms of ADRB1, ADRB2 and GNB3 subunit, no single nucleotide polymorphisms or haplotypes were associated with essential hypertension in the study of Filigheddu F et al., [79]. In the same study in females only, GNB3 SNPs and haplotypes were associated with the blood pressure response to beta blockers. On the other hand, in the study of Beige et al., [80] the 825T allele of this polymorphism was associated with 50% increased chance of having hypertension compared with non-T carriers.
An increase of plasma catecholamines that accompanies the essential hypertension suggests catecholamine synthetic enzymes as hypertension candidate genes. The locus for TH, the rate limiting enzyme in the biosynthesis of catecholamines, was found to be associated with essential hypertension in case-control study [81]. The group of Nielsen et al., investigated the relationship between a common SNP in the proximal TH promoter (C-824T) and blood pressure in a large general population sample, characterized by 24-h ambulatory blood pressure monitoring and office blood pressure measurement [82]. They reported that the C-824T SNP in the proximal TH promoter influences blood pressure and prevalence of hypertension in the general population.
GTP cyclohydrolase (GCH1) as a rate limiting enzyme of BH4 biosynthesis, is a cross-talk component for cateholamine and NO synthesis, both being crucial components of hypertension pathophysiology. Zhang et al., confirmed that the SNP C+243T affected both diasolic and systolic blood pressure, principally in females providing an important clue that both traits (NO secretion and autonomic activity) are jointly genetically determined, and that this fact can have important impact on future diagnosis, prognosis and treatment of hypertension [22]. These results, obtained on Caucasian population, remain to be confirmed on other populations.
Elevated levels of DBH, an important enzyme of catecholamine synthesis, are found in patients with essential hypertension. A novel polymorphism -1021C/T was found to interact with fasting plasma glucose level in association with hypertension, pointing on important gene-environment interaction in the genesis of hypertension obtained on Japanese population [83]. Another polymorphism of DBH C-970T was found to be associated with essential hypertension. These polymorphisms confers a wide range of autonomical traits: DBH expression and catecholamine secretion, heritable environmental stress-induced blood pressure increments, and ultimately – the risk for hypertension. Special attention deserves the observation that the C allele promoter variant, the most common variants in each population studied, is in line with “common disease/common allele” hypothesis for frequent pathophysiological traits that is certainly the case of essential hypertension. These findings are completed by the results on DBH polymorphism C-2073T on transcriptional and biochemical changes including augmented DBH secretion, eventuating in elevation of basal blood pressure, and hence cardiovascular risk [84]. This group in systematic and integrative approach revealed the role of DBH polymorphisms in the genesis of essential hypertension.
Methods in comparative genomics identified a genetic locus associated with blood pressure regulation in the stroke prone spontaneously hypertensive rats in a conserved synthetic group, corresponding to the gene for PNMT in humans [85]. A case/control association study was conducted to evaluate the relationship between genetic variants of the PNMT gene and risk for essential hypertension [86]. Two SNPs in the promoter region of the gene were genotyped, PNMT-148 and PNMT-353, in three ethnic samples: African American, American white, and Greek white population. A significant difference in allelic frequency of SNP-353 between hypertensives (38.02%) and normotensives (27.35%) in African Americans (P =.019) was found; however, no significant differences were observed for this SNP for the other ethnic groups. No association was found with SNP PNMT-148 in any of the ethnic groups. These results suggest that genetic variants of PNMT may play a role in the development of essential hypertension in some populations. The population dependent role of PNMT polymorphisms in the genesis of hypertension was confirmed by Huang et al., [87] who found a protective effect of G-367A and G-161A 2-SNPs AA haplotype in Han Chinese population.
Renalase (gene name RNLS), an enzyme with monoamine oxidase activity, is implicated in the degradation of catecholamines (Figure 3). Two SNPs rs2576178 A > G and rs2296545 C > G were investigated for the association with hypertension and no association was found [88]. On the other hand, Buraczynska et al., found the association between rs2296545 and hypertension in patients with type 2 diabetes, as though the association was found between rs2576178 and rs10887800 and hypertension in renal failure patients [89,90]. The role of this enzyme in the hypertension genesis might be syndrome dependent.
COMT is an important enzyme of norepinephrine and estrogene metabolism and plays an important role in the regulation of norepinephrine plasma levels. COMT SNP -1187G>C was found to be associated with essential hypertension in Japanese men [91], while rs4680 was found to be associated with hypertension in
Norwegian hypertension patients [92] and Sweedish men [93]. This result for rs4680 was confirmed by Htun et al., in Japanese men, with strong impact of an environmental factor-energy intake-on this interaction [94].
ALDH plays an important role in the interplay of catecholamine and alcohol metabolism. The association of this genotype with blood pressure was investigated in Chinese older men [95]. The association was found for alcohol dehydrogenase polymorphism but not for ALDH polymorphisms. This result speaks for the role of ethanol in the genesis of alcohol-associated hypertension, less for the role of aldehyde.
Even though there are novel data on the intravesicular interaction between H+ vesicular proton pump, CgA-catestatin system, catecholamine release and hypertension risk (in vitro, [96], no association study was done on the effect of SNPs of the vesicular proteins with hypertension.
In conclusion, it is worth to emphasize that hypertension, as the multifactorial multigenic disease, is strongly dependent on the cardiovascular reactivity to stress which is genetically determined and regulated by autonomic nervous system [97]. It is obvious that this gene-environment interactions are population dependent and that the molecular patterns of the cardiovascular reactivity to stress could also be population dependent. Still, this massively present disease in the developed countries speaks for the impact of environmental factor (stress) through different molecular “entrance gates” initiating the pathophysiological process that ends with common final phenotype- essential hypertension. This complex gene-environment interaction has to be addressed by systematic experimental approach combining gene, in vitro and in vivo studies of cardiovascular autonomic traits in different populations [22].
Cardiac necrosis and cardiomyopathy
Stress cardiomyopathy (SC) (tako-tsubo, left ventricular apical ballooning syndrome) is a newly reported condition afflicting older women, characterized by acute left ventricular systolic dysfunction, triggered by emotionally and physically stressful events, and occurring without significant coronary obstruction. Sympathetic nervous system hyperactivity has been implicated in the pathophysiology of SC[13]. Other forms of cardiomyopathy, like idiopathic dilatatory cardiomyopathy are also closely related to sympathetic overactivity [98].
SNPs involving the ADRB1 (amino acid positions 389 and 49) and ADRA2C (deletion 322-325) were investigated for susceptibility to SC [99]. Genotype polymorphism frequencies for these receptors were not significantly different in SC patients compared to female controls. These data suggest that these adrenergic polymorphisms do not mediate the sympathetic nervous system hyperactivity in SC patients. Pre-synaptic ADRA2 are essential feedback regulators to control the release of norepinephrine from sympathetic nerves. In the study of Regitz-Zagrosek et al., they tested whether a deletion polymorphism in the human ADRA2C gene (Del322-325) affects progression of HF in patients with dilated cardiomyopathy [100]. The results have shown that ADRA2C Del may be a novel, strong, and independent predictor of reduced event rates like death and implantation of a left ventricular assist device in dilated cardiomiopathy patients. The same type of cardiomyopathy was investigated for the effects of ADRB variants for Ser49Gly and Arg389Gly in the ADRB1; the 5' LC Arg19Cys, Arg16Gly, Gln27Glu, and Thr164Ile in the ADRB2; and Arg64Trp in the ADRB3 [101]. The Gly49 allele in the ADRB1 and the 5' LCCys19, Arg16, and Gln27 alleles in the ADRB2 were associated with a lower risk of HF in idiopathic dilated cardiomyopathy, suggesting that the ADRB1 and ADRB2 genes are modifier genes. Spinelli et al., postulated that polymorphisms associated with different ADRB responsiveness might play a role in the risk of SC, thus allowing the identification of those patients that are prone to SC after an adrenergic surge [102]. These authors confirmed the association of GRK5 L41polymorphism with SC, while the association for ADRB1, ADRB2 and GNAS1 were not found. This finding is consistent with the fact that GRK5 L41 polymorphism enhances desensitization and impairs ADRB response, reducing in this way the inotropic effect of catecholamines and compromising coronary blood flow [102]. On the other hand, the role of ADRB1 and ADRB2 in the genesis of SC was confirmed by Vriz et al., [103]. In one of the largest cohorts of SC in the world, Figtree et al., on the other hand found no association between ADRB1, ADRB2, GRK5 and COMT polymorphism and SC. Inhomogeneous results are possibly due to the racial, ethnic and sample size differences [104]. Also, different gene-environmental interactions can play an important role for the diversity of the obtained results.
There are several other neurocardiologic disorders where an abnormal catecholaminergic function is etiologic. For their role in the understanding of autonomic nervous system pathophysiology, special emphasis is drawn on:
a.Familial dysautonomia
b.Parkinson disease
cBaroreflex failure
d.Postural tachycardia syndrome
e.Catecholaminergic polymorphic ventricular tachycardia [13]
a.Familial dysautonomia (FD, Riley-Day syndrome), is one of several forms of hereditary sensory and autonomic neuropathies. This syndrome generally results from derangements in maturation and migration of neural crest-derived progenitor cells in the formation of sensory and autonomic population. The disease is transmitted as autonomic recessive trait, with the locus of genetic mutation on chromosome 9q31 which causes the deficiency of IKAP protein [105]. FD produces complex symptomatology, suggesting deficient activities of multiple neurotransmitter systems, catecholaminergic system being one of them. Elevated level of L-DOPA:DHPG ratio in patients [106] reflects decreased ability to synthesize NE and increased TH activity and depleted vesicular NE stores. This neurochemical pattern does not correspond to any single abnormality of catecholamine synthesis, signaling or metabolism, but suggest global arrested differentiation of catecholamine systems [13], mechanisms of which remain poorly understood.
Parkinson disease (PD) is often accompanied with cardiovascular dysautonomia. Catecholamine profile corresponds to decreased or absent synthesis, neuronal uptake and turnover. The findings are consistent with the loss of cardiac, extra cardiac sympathetic nerve terminals and baroreflex failure that progresses with time [107]. These results point on PD as not only central but also peripheral catecholaminergic neurodegeneration [13], where elucidation of cardiovascular dysautonomia could be the key point in understanding the pathogenesis of PD. An important point in understanding neurotoxicity in PD is the decrease of vesicular uptake: impaired vesicular sequestration builds up cytosolic catecholamines, which are toxic, by spontaneous auto-oxidation to quinines, enzymatic conversion to catecholaldehydes and by cytosolic catecholamine interactions with calcium and alphasynuclein (Figure 3). Still, no association was found in the study of VMAT2 polymorphism with PD in Japanese patients [108], as well as for the molecules participating in phosphorilation of alphasynuclein [109]. SNPs of other molecules in the catecholaminergic signaling and metabolic pathways play a significant role in the pathogenesis of PD, but the systematic review of this subject is out of scope of this review and can be found elsewhere [110].
c.Baroreflex failure is the syndrome usually with underlying organic pathology (familial paraganglioma syndrome, history of neck irradiation, surgical section of glossopharingeal nerve or NTS lesion) or with idiopathic etiology [13]. It is characterized by decreased/absent bradicardia responses to phenylephrine, decreased/absent tachycardia responses to nitroprusside, labile hypertension and orthostasis- or emotion-evocable episodes of marked increase in plasma catecholamine levels and blood pressure. The studies of individual/group genetic polymorphisms of catecholaminergic signaling and metabolic pathways in baroreflex failure are still lacking.
d.Postural orthostatic tachycardia syndrome (POTS) is characterized by a sustained increase in heart rate (30-120bpm) within 10min of orthostasis or head up tilt in the absence of orthostatic hypotension [111]. POTS include also exercise intolerance, fatigue, chest pain and dizziness. It is considered as “partial dysautonomia”, manifesting incomplete, mild autonomic dysfunction that compensatorily activates sympathetic drives to other regions via baroreflex mechanisms. Nickander et al., [112] found no association between SNPs of ADRB2, ADRA2C, NET and mitochondrial complex I. Even though there was not confirmed the strong association between DBH genotype and POTS, Garland et al., suggest that differences in DBH activity in POTS patients might reflect the different levels of sympathetic activation [113]. In children with POTS of Japanese ethnicity an association was found with GNB3 polymorphism while the GNAS1 polymorhism was not associated with the syndrome [114]. Different results on the role of genetic polymorphisms of catecholaminergic signaling and metabolism might be the consequence of different sample size, age and ethnicity. More, the data on the influence of other SNPs of genes of catecholaminergic molecular machinery are lacking.
e.Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an ion channelopathy of genetic origin (mutation of cardiac RyR2 and CASQ2 gene, Figure 1) characterized by life threatening ventricular arrhythmias triggered by sympathetic activity (physical activity or emotional stress) [115]. The identification of genes underlying stress-induced life-threatening arrhythmias in intact heart revealed that different arrhythmias are the same disease with different phenotype expression, all of them potentially leading to the clinical entity-SCD. RyR2 and CASQ2 are molecules on the crossroad between the mechanisms of cardiac electrical and mechanical activation, having in this way an important role in the genesis of cardiomyopathy, another catecholamine associated disease [13]. The systematic analysis of the phenotypes with enhanced adrenergic response to physical or emotional stress in patients with calcium channelopathies is still lacking.
Vagally mediated cardiovascular diseases
Candidate genes for vagally mediated cardiovascular diseases are, as the ones for sympathetically mediated disorders, all along the vagal signaling and metabolic pathway (Figure 2). These are the genes for enzyme of biosynthesis of acetylcholine (Ach): choline acetyl transferase (CholAT), vesicular Ach transporter (VACHT), enzyme of Ach degradation acetylcholine esterase (AchE), receptors (muscarinic, M1-M5 and nicotinic, N), G proteins (pertussis toxine sensitive inhibitory G protein Gi and pertussis toxine insensitive G protein Gq), enzymes of intracellular signal transduction (adenyl cyclase- AC, guanylil cyclase- GC, protein lipase Cβ (PLCβ), protein kinase C (PKC), key enzyme in endothelium dependent vascular smooth muscle relaxation nitric oxide synthase (NOS), and ion channels, with special emphasis on Ca2+ and K+ channels.
Vasovagal syncope
Vasovagal syncope (VVS, reflex syncope, neuraly mediated syncope, neurocardiogenic syncope, emotional fainting) is a common clinical problem, characterized by transient episodes of loss of consciousness due to abnormal autonomic activity [111,116,117]. Classical VVS is mediated by emotional, and/or orthostatic stress. It is diagnosed when precipitating events such as fear, severe pain, emotional distress or prolonged standing are associated with typical prodromal symptoms [118]. Pathophysiology of VVS is still unclear [116,118-122]. According to the polyvagal theory, the neural regulation of the autonomic nervous system passes through three global stages, each with associated behavioral strategy [123]. In mammals a dorsal vagal complex, the vestigial immobilization system, has a role in generation of VVS, as a typical behavioral response to environmental challenges. This theory puts the light on the possible pathophysiological mechanisms of centrally (emotionally) induced VVS, while the VVS induced by orthostatic stress has a variety of proposed pathophysiological mechanisms, where vagal system again plays pivotal role [116]. Hereditary aspects of VVS were studied in 19 studies so far [124]. Clinical descriptions of familial VVS are scarce and determined a positive family history in 19-90% of the VVS patients [125-127].
Association studies of neurohumoral candidate genes and VVS
Few studies examined potential genetic polymorphisms associated with VVS and, due to still unrevealed pathophysiological process, were focused on different neurohumoral systems that participate in the regulation of circulatory homeostasis.
Newton et al., examined the influence of ACE insertion/deletion (I/D) polymorphism, which could lead to altered circulating angiotensin levels, finding that there was no significant difference in distribution of ACE I/D gene frequencies in 165 cases compared with a large national control population [128]. The endothelin system was also investigated for the role of endothelin-1 genetic polymorphisms in VVS, and its association with vasodepressive form of VVS was found [129].
The functional role of ADRB1 polymorphism Arg389Gly on susceptibility to develop a positive head up tilt test (HUT) was suggested [130]. Subjects with Gly389 have lower cardiac inotropic response to catecholamines and this could therefore dysregulate the autonomic respose to HUT [130]. Marquez et al., [117] studied this polymorphism in 50 patients with unexplained syncope. The Gly389 allele was more frequently present in VVS patients with positive HUT test than in HUT negative subjects [117]. Hernandes-Pacheco et al., [131] also investigated the correlation between VVS and ADRB1 polymorphism at the 389 position. Their results suggest that the VVS has a genetic component associated with Arg389Gly polymorphism of ADRB1. Sorrentino et al., considered eight polymorphisms influencing sympathetic activity as a major risk factor for VVS in Italian patients [132]. They analysed polymorphisms of ADRA1A, ADRB1, ADRB2, GNB3, DBH and SLC6A4 gene. No association of the aforementioned genetic variants with both HUT outcomes and new syncopal episodes during follow-up was found [132]. Considering the small number of patients that were enrolled in these studies, further studies are needed to correlate haemodynamic resposes to autonomic tests with polymorphic variations in genes. Even though some data speak for the role of ADRB1 in susceptibility to VVS [117], there is an increasing evidence that genetic polymorphisms of sympathetic signaling pathway are not associated with tiltinduced VVS [128]. This brought the authors to the conclusion that an emphasis should be put on the parasympathetic genes and their interaction with the environment.
Studies of candidate genes of acetylcholine homeostasis: biosynthesis (CholAT), vesicular packing (VACHT), enzyme degradation (AchE) and choline uptake from synaptic cleft (ChT) in VVS
Even though there is a considerable amount of the literature that speaks for the role of CholAT in Alzheimer disease [133], bipolar disorder [133,134] and schizophrenia [135] to our knowledge no study was done on the role of its genetic polymorphisms in VVS.
Due to the key role of Ach in locomotion it was very complex to study on animal models the role of genetic disturbance in cholinergic neurotransmission of autonomic nervous system and consequently on cardiovascular system [136,137]. Recently, a mutant animals for different components of cholnergic signaling pathway have been developed [138], which opened the trace of studying the role of cholinergic genes in initiation and, interestingly, the withdrawal of the pathophysiological process of different diseases of autonomic nervous system[139]. The cardiovascular parameters (heart rate and arterial blood pressure) important for the pathophysiological process of VVS, were not altered in VACHT mutant mice, while the sympathetic predominance on the heart level was observed. Still, an important caveats for these results are that the measuring of the cardiovascular parameters was performed under the halothane anesthesia, which can independently alter neural regulation of cardiocerebrovascular system [140,141]. The essential result on molecular genetics level of this complex and interesting study is the evidence that the insufficiency of acetylcholinergic neurotransmission in VACHT mutant mice induce the cascade of genetic expression alterations of the vast number of proteins participating in autonomic nervous system regulation: M1, M2 and M3 receptors, ADRB1, GRK2 and GRK5. This is in line with the increasing evidence that some neurocardiovascular diseases like HF [142,143] and VVS [111]are the result of complex interactive dysfunction of both sympathetic and parasympathetic branch of autonomic nervous system that occurs on systemic and molecular level [6,13]. The role of VACHT was investigated in different addictive behavior [144], locomotion [145] and cognition [146], and it’s role in autonomic dysfunctions was also observed: in HF [137], hypertension and cerebrovascular disease [147]. The fact that VVS is a behavioral phenomenon with emphasized autonomic phenomenology speaks indirectly in favor for the possible role of VACHT in VVS pathophysiology, and plausibly to it’s genetic background. AchE was found to be an important factor of the VVS and its cardioinhibitory component [148], taking into account vagal pathophysiology interacting with vasodepressor mechanisms. A cholinergic disfunction on the level of CholAT, VACHT and ChT gene transcription were described for the central cholinergic dysfunction in Huntington disease [149], but due to the inaccessibility of peripheral vagal fibers as well as to the fact that animal models of VVS are still developing [137,150], this issue is still not assessed in VVS. More, though there is prevailing opinion that VVS is the pathophysiological entity where multiple gene-gene and gene-environment interaction should be taken in the consideration [129], there is still a lack of data that include the parasympathetic genes in this network. In line with this hypothesis, recent data propose an animal model of vagal hyperactivity [137], assessed by relatively noninvasive technique of heart period time domain analysis [6], suggesting that inheritance model of vagal hyperactivity could correspond to polygenetic model with the partial sex-limited character. New investigations in the area of genetic studies are needed for the confirmation of these hypotheses on the role of Ach homeostasis gene in VVS.
Studies of muscarinic receptor candidate genes and VVS
An interesting association was found between Alzheimer diseases (pathology with central Ach dysfunction) and VVS (pathology with peripheral acetylcholine dysfunction), with the crucial role of muscarinic receptors (M1) [151]. Central muscarinic receptors play an important role in different cardiovascular reflexes, like Bezold-Jarish reflex and baroreflexes [151].Thus, the alterations in central M1 activity can cause the autonomic dysfunction of cardiovascular system and makes it an interesting gene-candidate for the association of different gene polymorphisms and the incidence of VVS. The positive effect of ipratropiumbromide in the therapy of central VVS points out that the muscarinic receptors play an important role in the pathophysiology of VVS [152].
Novel data on the genetic overexpression of M2 and M3 receptors in the states of vagal hyperactivity, as it is the case of VVS, suggest that the existence of SNPs of these receptors (83% of vagally hyperactive animals) has an important role in the genesis of this pathophysiological state [153]. A compensatory increase of AchE gene expression in the same animal model of VVS speaks for the important gene-gene interaction in the acetylcholinergic signaling pathway. The association studies of SNPs of muscarinic receptors and VVS are lacking, but the growing corpus of genetical investigation on the role of muscarinic receptors in VVS will open the way for these investigations.
Association studies of G-proteins candidate genes and VVS
Lelonek et al., studied 217 VVS patients free from any other disease that were examined related to HUT results and genotyped for polymorphism in GNAS1 gene (polymorphism C393T), GNB3 (polymorphism C825T) and for the cardiac regulator of G-protein signaling RGS2 (polymorphism C1114G) [154]. In multivariate logistic regression analysis, the homozygotes 825TT GNB3 (OR 0.37; 95% CI 0.14-0.97; P < 0.05) and body mass index (OR 0.87; 95% CI 0.78-0.97; P = 0.005) were independently associated with a lower chance of positive tilting results. The same authors studied in 68 positive tilted patients the GNB3 C825T polymorphism, which was associated with increased G proteinmediated signal transduction [155]. On the basis of their results, the predisposition to VVS seems not to be associated with the GNB3 C825T polymorphism. The frequency of GNB3 825T allele was significantly higher in patients with non-typical VVS history than in group with typical history. Lelonek et al., investigated the relationship between GNAS1, GNB3 and the RGS2 with Calgary syncope syndrome score as the potential biomarkers for screening test for VVS [156]. Their results showed that 393T GNAS1 remained independent factor associated with positive tilt results. The authors suggested the variation of GNAS protein gene as a helpful test for identifying tilt-induced VVS patients.
Although neurogenetic influences on the occurrence of VVS episodes seem possible, specific high risk polymorphisms have not been identified so far. Differences in vasovagal responses between the subjects and different mechanisms for the development of VVS suggest that various genetic polymorphisms could be of significance.
Studies of other intracellular molecular signaling pathway genes and VVS
Increased concentration of plasma cAMP, implying on activity of AC has a role in pathophysiological process of VVS [157]. The authors link this result to the increased activity of sympathetic nervous system preceding the syncope event, though the contribution of parasympathetic signaling route cannot be excluded. To the best of our knowledge no investigation on the role of AC genetic polymorphisms in VVS has been done.
Calcium is one of the most important intracellular signaling particles in the parasympathetic signaling pathway. Dysfunction of calcium homeostasis is recognized to play an important role in many cerebrovascular and neurodegenerative diseases. Abnormalities of voltage gated calcium channels, the most important regulators of intracellular calcium concentration participate in vast number of human diseases, like migraine and episodic ataxia [158]. As parasympathetic activity plays a certain role in generation of these diseases[159], it is reasonable to hypothesize that genetic modulation of voltage gated calcium channels could play a role in pathophysiological process of VVS. So far, no studies were performed on the role of voltage gated Ca2+ channels genetic polymorphisms in VVS.
Adenosine, an ubiquitous purinergic nucleoside present in all living cells, participates in vast number of physiological processesregulation of vascular smooth muscle tone, heart rate and neurotransmission. It’s characteristic that, when administered exogenously, provokes negative chronotropic and dromotropic effects, was proved useful as a provocative test for VVS [160]. The mechanisms of it’s syncopogenic actions are still unexplained [161]. An interesting point is that the purinergic receptors on myocardial cells share common signaling output with muscarinic receptors-opening the potassium channels (Figure 3) [162], implying that both physiological regulator systems of cardiomyocyte excitability, purinergic and cholinergic, could have the common bottleneck in VVS - the K+ channels. Differently from well known K+ channelopathies like long QT syndrome [163] and short QT syndrome [164], there are no data about the role of genetically modulated K+ channels in the pathogenesis of VVS. Coming back to the role of adenosine in VVS, the data on potential role of the genetic modulation of protein components of adenosine numerous biosynthetic pathways and the pathways of degradation [165] is still lacking.
Candidate genes in vagally mediated atrial fibrillation
Atrial fibrillation (AF) affects more than 5 million people worldwide, making a prevalence of 1% in the general population. It is characterized by rapid, erratic electrical activation of the atrial myocardium, resulting in the loss of effective contractility an increased likelihood of clot formation, and an increased risk of stroke. AF is presented as familial and non familial form [166].
Some studies investigating a molecular pathogenesis of AF have focused on familial forms of the disease. Recently, a mutation in KCNQ1, a K+ channel gene also implicated in a form of the long- QT syndrome, was identified as the molecular basis of autosomal dominant AF in a single family from China [167]. Two additional families with autosomal dominant AF have been identified and the loci mapped to 10q22–q24 and 6p14–p16 [168,169]. However, a causal gene has not been identified. Familial forms of AF may provide insight into the molecular pathways involved in these selective cases of the disease, but the genetic defects identified may not be representative of the pathogenesis in the more common, nonfamilial form of AF.
Differently from monogenic forms, caused by rare genetic mutations with high penetrance, the more present form of AF is likely to be caused by common genetic polymorphisms that arrhythmogenically interact with environmental factors [170]. A vast number of different gene polymorphisms were investigated and, interestingly, a morphological remodeling of different proteins due to genetic modulation gave the same AF form as did electrophysiologically remodeled proteins in different patophysiological conditions accompanied by AF. This speaks in favor of the common underlying mechanisms, that are still insufficiently revealed [171], with fact that different mechanisms operate in different circumstances [172].
Being the focus of this review, we will consider the balance between sympathetic and vagal influences as an important predictor of AF. Vagal predominance has been observed in the minutes preceding the onset of AF in patients with idiopathic paroxysmal AF, while in patients with organic heart diseases there is a shift toward sympathetic predominance [173]. In general, vagally mediated AF occurs at night or after meals [174], while adrenergically induced AF typically occurs during the daytime. In patients with vagally mediated AF, the more common form, adrenergic blocking drugs or digitalis sometimes worsen symptoms [175]. Some results suggest that simultaneous sympathovagal discharge is profibrillatory [176-178]. These conflicting results suggest that the complex interaction between autonomic nervous system and AF is due to individual variability, with some patients having more emphasized autonomic triggers than others [179].
Studies of candidate genes of acetylcholine homeostasis: biosynthesis (CholAT), vesicular packing (VACHT), enzyme degradation (AchE) and choline uptake from synaptic cleft (ChT) in vagally mediated atrial fibrillation
Cholinergic neurons, both central and peripheral, express three proteins-CholAT, VACHT and ChT, that are in physiological equilibrium and in the function of the maintenance of physiological levels of Ach. The interrelation of the genes of these proteins and their polymorphic variances was investigated in syndromes like congenital myasthenic syndrome with episodic apnea [180] and genetically modulated animal models of central cholinergic dysfunction [181]. Even though it is intuitive that the gain-of-function genetic modulations influencing the quantity and the release of Ach in neural-cardiomyocite synaptic cleft is of interest for pathophysiology of vagally mediated AF, no association studies have been performed in this direction.
Studies of muscarinic receptor candidate genes and vagally mediated AF
Increase of Ach release to neural-myocardiocyte synaptic cleft increases cardiac vulnerability to AF by muscarinic receptor-mediated shortening of refractory period. Different antiarrhythmic drugs perform their action by inhibiting muscarinic Ach receptors, but also by inhibiting the G proteins, the transduction proteins of cholinergic signaling pathway, and, finally, directly acting on K+ channels [182]. This fact makes these proteins an important candidate for genetic evaluation to vagally induced AF susceptibility.
The importance of muscarinic receptors in age dependent increase for AF susceptibility is strengthened by the fact that the expression of M2 in atrial cells is region dependent [183] and increases with age [184].
The data from the studies of systemic interrelations of sympathetic and parasympathetic system [185] speak for the importance of the adequate dynamic interrelation of these systems in different physiological states where cardiac autonomic regulation is under specific strain, like sleep [11,141,185] or positional and pharmacologic modulation of autonomic nervous system [186]. Having on mind that muscarinic receptors play an important role both for the downward transmission of cholinergic signal as well as for inhibition of adrenergic signaling [187,188] (Figure 2) it is probable that its functional modulation due to genetic variability might be of importance for initiation of different arrhythmogenic episodes, AF being one of them.
Association studies of G-proteins candidate genes and vagally induced AF
G proteins, the cellular transduction proteins, are of particular interest in the pathophysiology of AF. G proteins can couple both with M2 and ADRB2 receptors and mediate in this way the opposing effects of parasympathetic and sympathetic nervous system on the Ca2+ and K+ ion currents and cardiomyocite excitability [189]. The polymorphisms of GNB3 (C825T substitution), initially identified as a risk factor for hypertension [190] was targeted for its association with atrial fibrillation [191]. Although previously identified as a cardiovascular risk factor, in the case of atrial fibrillation TT genotype manifested protective effect.
Magnitude and duration of G protein coupled receptor responses are regulated by the activity of the RGS family of proteins [189]. These proteins act as GTPase stimulating proteins of GNAS1. Many of these proteins are detectable in sinoatrial node, atrioventricular node, atrial and ventricular cells and perform there their modulating action on cellular electrophysiology. Two RGS proteins, RGS6 and RGS4, are found to be critical regulators of M2 transmission, functioning as the brake on vagal activity. Since there are results on the role of RGS proteins in AF in knock out animals [192], it is intuitive to conclude that the genetic polymorphisms of RGS proteins in the sence loss-of –function might be of importance in the pathophysiology of AF. Even though the research on the role of RGS polymorphisms in VVS [155] and hypertension [193,194] has been done, no studies on their role in AF has been performed.
Studies of other intracellular molecular signaling pathway genes and vagally induced AF
Even though ion handling molecules in AF can be observed independently, they make an integral, conclusive part of the neural signaling pathway to cardiomyocite [195-197]. In order to follow the scope of this review, we put an emphasis only on the most important findings in the genetic research of these molecules that participate in neural signaling dysfunction. For more detailed review on the ion handling molecules in AF the paper of Mahida and collaborates should be addressed [198].
Delayed afterdepolarizations are one of the basic mechanisms of AF generation, which results from altered intracellular homeostasis of Ca2+ [170] (Figure 2). The principal Ca2+ handling molecules are voltage-dependent Ca2+ channels, ryanodine receptors on sarcoplasmatic reticulum (RRS), the sarcoplasmatic Ca2+ ATPase (SERCA) and the cell membrane Na+, Ca2+-exchanger [199].
A feed-forward relation between AF and gene expression of above listed Ca2+ handling molecules was observed [195-199]. Additionally, the association was found between the large genomic deletion in RRS gene and different arrhythmic heart diseases that include AF [200]. A heterozygous missense mutation in RRS gene is also associated with different arrhythmic heart disease [201]. A role of RRS gene mutation in the AF genesis was confirmed in animal models, too [202].
The first link between ion channelopathies and AF was provided by Chen and collaborates [167]. The KCNQ1 gene encodes a pore-forming alpha subunit which associates with beta subunits, forming a delayed-rectifier K+ current which is prominent at higher heart rates and adrenergic stimulation during the late phase of the action potential. This mutation causes a marked slowing of K+ channel deactivation. A missense polymorphism in the minK gene is found to be associated with AF in patients from Taiwan population [203].
The one of genetic backgounds of AF was a loss-of-function SNP in KCNE1 which encodes the slow delayed-rectifyer K+ channel beta-subunit mink [204].
The mutation of KCNA5 that disrupts the structure and the function of Kv.1.5 conferees the susceptibility to action potential prolongation and the generation of AF [205]. A SNP of the KCNN3 gene encodes dysfunctional Ca2+ activated K+ channel and is associated with AF [206].
Experimental data demonstrated that NO is involved in cardiac vagal activity, the inhibition of sympathetic activity, and the atrial electrophysiological remodeling [207], suggesting the potential pathophysiological route from vagal activity down to the heart electrophysiological remodeling. The authors evaluated the role of eNOS -786T>C, 894G>T, 4a/4b and of minK S38G polymorphisms as predisposing factors to non-valvular AF. The minK 38G allele was significantly associated with susceptibility to non-valvular AF. The eNOS -786C allele weakly influenced non-valvular atrial fibrillation, according to the dominant model (OR=1.50, P=0.01). These findings suggest a role for minK and eNOS genes as predisposing factors to non-valvular AF.
These data speak in favor of the fundamental role of genes coding proteins of Ca2+ and K+ homeostasis in cardiac arrhythmogenesis, but also imply that the change at the functional level of these genes influences arrhythmogenic potential of the whole heart, where vagally induced AF represents only a segment of the complex picture of cardiac arrhythmias.
Conclusions
The scope of this review was to present the state of the art on the role of candidate gene polymorphisms in signaling chain of neural transmission and metabolism in neurocardiovascular diseases, enlightening the missing links of data on this issue.
In these multifactorial and prevalently multigenic diseases emphasize is put on the impact of neural (dys)regulation on the global pathophysiological situation. This impact is sometimes etiological factor and sometimes precipitating factor for aggravation of symptoms, in the worse case the fatal outcome [13]. From proteo-genetic point of view a vast number of molecules all along the neural signaling and metabolic pathways can be functionally modified and consequently be the background of the predisposition to the disease, its faster progression or its complicated form. Consequently, the identification of these genes makes an excellent diagnostic tool in the sense of right-time and right-choice therapy. The thorough evaluation of the impact of genetically modulated classical molecules in neurocardiovascular communication is still missing, while on the other hand there is an increasing knowledge on new molecules with modulatory effect on these systems.
Special emphasis should be put on the fact that the impact of neural dysfunction on autonomic nervous system diseases of CV system is rarely the consequence of isolated dysfunction of parasympathetic or sympathetic nervous system, but more of the disequilibrium in joined modulation and dynamics of these two systems: this is the case for HF [142,143], AF [176-178] and VVS [13,6]. That is why, for the future neurogenetic research, an emphasis should be put on signaling molecules that provide the cross-talk between these two systems: on intercellular level (i.e. muscarinic receptors), on intracellular level (G proteins and molecular machinery that regulates their activity), or on the level of the integration of signaling process, like Ca2+ handling molecules or K+ channels. This also speaks strongly for the future necessity of redefining pathophysiological syndromes and didactic classifications of CV dysautonomias.
The vast palette of data obtained from association studies between SNPs of autonomic nervous system and different pathophysiological entities picture the genetic plasticity of neurocardiovascular signaling pathways. Different external or internal factors could act on genetic structure and trace different functional pathways of the same neurocardiovascular phenotype/disease [66,86,97]. This is typically the case of essential hypertension.
Additionally, an aggregation of positive results for the associations between certain genes and different pathophysiological phenotypes could be observed, pointing on the specific "neurocardiovascular genetic hotspots", obviously the genes with high pleiotropy. This is the case for: ADRB2 polymorphism association with hypertension [63], HF [48,49], SCD [208] and CAD [209], and G protein molecular machinery- GNAS1 association with hypertension [76], HF [210] and VVS [156] and RGS association with VVS [155] and hypertension [193,194]. Having on mind that many neurocardiovascular diseases occur in clusters [210] identification of these genes could be of particular interest as a diagnostic tool in complex forms of these diseases.
The opening gate of GWAS studies will offer an insight into new candidate genes and new potential pathophysiological pathways that might elucidate better the neural role in the pathophysiological process of the neurocardiovascular disease. An important interpretational gaps that exists between the level of gene functioning, the cellular level of electrophysiological functioning and, finally, systemic physiological and the behavioral level of functioning of the organism, should be overcome by combined genetic, in vitro and in vivo studies of autonomic traits [211]. Additionally, due to the feed forward modulating effect of the pathophysiological process on the gene level, it is usually difficult to establish cause-effect relation [212]. Consequently, the creative unification of the insight from the systemic level of the disease (clinically-the stage of the disease) with the insight of data obtained by modern genetic techniques will trace the way for new interpretations of the interaction between genes and pathophysiological process in neurocardiovascular diseases. Syndomes like POTS, CPVT or cardiac dysautonomia in PD are indicative “intermediate phenotypes” that can offer both to the researchers a novel aspects on genetic background of ANS diseases and to clinicians a valuable landmark for disease progression. Role of the molecules of parasympathetic homeostasis [204-207] in genesis and regression of certain neurocardiovascular disorders is on the very beginning of the promising future SNPs and GWAS studies.
Drugs that interfere with autonomic control of the heart and vasculature that are used in the treatment of arrhythmias, hypertension, and HF are still not able to cure the diseases [6]. New data obtained from neurogenetic approach will improve the disease outcome by pharmacologic, gene and behavioral modulationof the autonomic nervous system [66,213,214].
Acknowledgement
This work was financed by the Ministry of Education, Science and Technological Development of the Republic of Serbia, project III 41028. We thank to the reviewers for the constructive suggestions that improved the manuscript. Special thanks to Dr Esma Isenovic, Dr Boban Stanojevic and Dr Vesna Mandusic and for valuable scientific insights. We thank to Dr Dragan Alavanti? for critical overview of the manuscript.
6481
References
- Rowell, LB. Reflex control during orthostasis. In Rowell, L. B. (Ed.) Human cardiovascular control. 1993; Oxford University Press, inc. New York.
- Berne, R. M., et al., 1993; The cardiovascular system: regulation of heart beat.
- Franchini, K. G., et al., 1996. Autonomic control of cardiac function. In Robertson, D., Low, P. A. and Polinsky, R. J. (Eds.) Primer on the autonomic nervous system. San Diego, Academic Press, Inc.
- Hamill, R. W., 1996. Peripheral autonomic nervous system. In Robertson, D., Low, P. A. and Polinsky, R. J. (Eds.) Primer on the autonomic nervous system. San Diego, Academic Press, Inc.
- Bojic, T., Sudar, E., Mikhailidis, D., Alavantic, D., Isenovic, E. The role of G protein coupled receptor kinases in neurocardiovascular pathophysiology. Arch Med Sci 2012; 8: 970-977.
- Bajic, D., Loncar-Turukalo, T., Stojicic, S., Sarenac, O., Bojic, T., et al. Temporal analysis of the spontaneous baroreceptor reflex during mild emotional stress in the rat. Stress 2010; 13: 142-154.
- Bjelakovic, B., Ilic, S., Chouliaras, K., Milovanovic, B., Vukomanovic, V., et al. Heart rate variability in children with exercise-induced idiopathic ventricular arrhythmias. Pediatr Cardiol 2010; 31: 188-194.
- Silvani, A., Asti, V., Bojic, T., Ferrari, V., Franzini, C., et al. Sleep-dependent changes in the coupling between heart period and arterial pressure in newborn lambs. Pediatr Res 2005; 57: 108-114.
- Silvani A., Bojic T., Franzini C., Lenzi P., Walker AM., et al. Sleep-related changes in the regulation of cerebral blood flow in newborn lambs. Sleep 2004; 27: 36-4.
- Silvani, A., Bojic, T., Cianci, T., Franzini, C., Lodi, CA., et al. Effects of acoustic stimulation on cardiovascular regulation during sleep. Sleep 2003; 26: 201-205.
- Zoccoli G., Andreoli E., Bojic T., Cianci T., Franzini C., et al. Central and baroreflex control of heart rate during the wake-sleep cycle in rat. Sleep 2001; 24: 753-758.
- Goldstain, D. The Autonomic Nervous System in Health and Disease, New York, Marcel Dekker, Inc.
- Fisher, J.P., Paton, J.F. The sympathetic nervous system and blood pressure in humans: implications for hypertension. J Hum Hypertens 2012; 26: 463-475.
- Eisenhofer, G., Kopin I.J., Goldstein DS Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 2004; 56: 331-349.
- Weber, C., Noels, H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 2011; 17: 1410-1422.
- Rao, F., Zhang, L., O'Connor, DT. Complex trait genetics the role of mechanistic "intermediate phenotypes" and candidate genetic loci. J Am Coll Cardiol 2008; 52: 166-168.
- Silvani, A., Bojic, T., Cianci, T., Franzini, C., Lodi, CA., et al. Effects of acoustic stimulation on cardiovascular regulation during sleep. Sleep 2003; 26: 201-205.
- Silvani, A., Bojic, T., Cianci, T., Franzini, C., Lodi, CA., et al. Effects of acoustic stimulation on cardiovascular regulation during sleep. Sleep 2003; 26: 201-205.
- Middlekauff, HR., Park, J., Agrawal, H., Gornbein, JA. Abnormal sympathetic nerve activity in women exposed to cigarette smoke: a potential mechanism to explain increased cardiac risk. Am J Physiol Heart Circ Physiol 2013; 305: H1560-1567.
- Michalsen, A., Knoblauch, NT., Lehmann, N., Grossman, P., Kerkhoff, G., et al. Effects of lifestyle modification on the progression of coronary atherosclerosis, autonomic function, and angina--the role of GNB3 C825T polymorphism. Am Heart J 2006; 151: 870-877.
- Zhang L., Rao F., Zhang K., Khandrika S., Das M., et al. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest 2007; 117: 2658-267.
- Shinozaki, K., et al. Coronary endothelial dysfunction in the insulin-resistant state is linked to abnormal pteridine metabolism and vascular oxidative stress. J Am Coll Cardiol 2001; 38: 1821-1828.
- Farzaneh-Far, R., Desir, G.V., Na, B., Schiller, N.B., Whooley, MA. A functional polymorphism in renalase (Glu37Asp) is associated with cardiac hypertrophy, dysfunction, and ischemia: data from the heart and soul study. PLoS One 2010; 5: e13496.
- Happonen, P., Voutilainen, S., Tuomainen, .TP., Salonen, J.T. Catechol-o-methyltransferase gene polymorphism modifies the effect of coffee intake on incidence of acute coronary events. PLoS One 2006; 1: e117.
- Robador, PA., et al. Aldehyde dehydrogenase type 2 activation by adenosine and histamine inhibits ischemic norepinephrine release in cardiac sympathetic neurons: mediation by protein kinase Cepsilon. J Pharmacol Exp Ther 2012; 343: 97-105.
- Han, H., Wang, H., Yin, Z., Jiang, H., Fang M., et al. Association of genetic polymorphisms in ADH and ALDH2 with risk of coronary artery disease and myocardial infarction: a meta-analysis. Gene 2013; 526: 134-14.
- Wang, Q., Zhou, S., Wang, L., Lei, M., Wang, Y., et al. ALDH2 rs671 Polymorphism and coronary heart disease risk among Asian populations: a meta-analysis and meta-regression. DNA Cell Biol 2013; 32: 393-399.
- Prystowsky, EN., Padanilam, BJ., Joshi, S., Fogel RI Ventricular arrhythmias in the absence of structural heart disease. J Am Coll Cardiol 2012; 59: 1733-1744.
- Kanki, H., Yang, P., Xie, HG., Kim, R.B., George, AL Jr., et al. Polymorphisms in beta-adrenergic receptor genes in the acquired long QT syndrome. J Cardiovasc Electrophysiol 2002; 13: 252-256.
- Paavonen, KJ., Swan, H., Piippo, K., Laitinen, P., Fodstad, H., et al. Beta1-adrenergic receptor polymorphisms, QTc interval and occurrence of symptoms in type 1 of long QT syndrome. Int J Cardiol 2007; 118: 197-202.
- Ulucan, C., Cetintas, V., Tetik, A., Eroglu, Z., Kayikcioglu, M., et al. Beta1 and beta2-adrenergic receptor polymorphisms and idiopathic ventricular arrhythmias. J Cardiovasc Electrophysiol 2008; 19: 1053-1058.
- Wieneke, H. et al. Better identification of patients who benefit from implantable cardioverter defibrillators by genotyping the G protein beta3 subunit (GNB3) C825T polymorphism. Basic Res Cardiol 2006; 101: 447-45.
- Wieneke, H. et al. Polymorphisms associated with ventricular tachyarrhythmias: rationale, design, and endpoints of the 'diagnostic data influence on disease management and relation of genomics to ventricular tachyarrhythmias in implantable cardioverter/defibrillator patients (DISCOVERY)' study. Europace 2010; 12: 424-429.
- Schreieck, J., Dostal, S., von Beckerath, N., Wacker, A., Flory, M., et al. C825T polymorphism of the G-protein beta3 subunit gene and atrial fibrillation: association of the TT genotype with a reduced risk for atrial fibrillation. Am Heart J 2004; 148: 545-550.
- Adlam, D., Herring N., Douglas G., De Bono JP., Li D., et al. Regulation of β-adrenergic control of heart rate by GTP-cyclohydrolase 1 (GCH1) and tetrahydrobiopterin. Cardiovasc Res 2012; 93: 694-70.
- Uhl, GR., Li, S., Takahashi, N., Itokawa, K., Lin, Z., et al. The VMAT2 gene in mice and humans: amphetamine responses, locomotion, cardiac arrhythmias, aging, and vulnerability to dopaminergic toxins. FASEB J 2000; 14: 2459-2465.
- Parati, G., Esler, M. The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 2012; 33: 1058-1066.
- Liggett, SB., Wagoner, LE., Craft, LL., Hornung, RW., Hoit, BD., et al. The Ile164 beta2-adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J Clin Invest 1998; 102: 1534-1539.
- Liggett, SB. Pharmacogenetics of beta-1- and beta-2-adrenergic receptors. Pharmacology 2000; 61: 167-173.
- Borjesson, M., Magnusson, Y., Hjalmarson, A., Andersson, B. A novel polymorphism in the gene coding for the beta(1)-adrenergic receptor associated with survival in patients with heart failure. Eur Heart J 2000; 21: 1853-1858.
- Bristow, MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101: 558-569.
- Podlowski, S., Wenzel, K., Luther, HP., Muller, J., Bramlage, P., et al. Beta1-adrenoceptor gene variations: a role in idiopathic dilated cardiomyopathy? J Mol Med (Berl) 2000; 78: 87-93.
- Mason, DA., Moore, JD., Green, SA., Liggett, SB. A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J Biol Chem 1999; 274: 12670-12674.
- Tesson, F., Charron, P., Peuchmaurd, M., Nicaud, V., Cambien, F., et al. Characterization of a unique genetic variant in the beta1-adrenoceptor gene and evaluation of its role in idiopathic dilated cardiomyopathy. CARDIGENE Group. J Mol Cell Cardiol 1999; 31: 1025-1032.
- Mialet Perez, J., Rathz, DA., Petrashevskaya, NN., Hahn, HS., Wagoner, LE., et al. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 2003; 9: 1300-1305.
- Small, KM., Wagoner, LE., Levin, AM., Kardia, SL., Liggett, SB. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med 2002; 347: 1135-1142.
- Brodde, O.E., Büscher, R., Tellkamp, R., Radke, J., Dhein, S., et al. Blunted cardiac responses to receptor activation in subjects with Thr164Ile beta(2)-adrenoceptors. Circulation 2001; 103: 1048-1050.
- Pereira, SB., Velloso, MW., Chermont, S., Quintao, MM., Nunes Abdhala, R., et al. β-adrenergic receptor polymorphisms in susceptibility, response to treatment and prognosis in heart failure: implication of ethnicity. Mol Med Rep 2013; 7: 259-265.
- Shin, J., Lobmeyer, MT., Gong, Y., Zineh, I., Langaee TY., et al. Relation of beta(2)-adrenoceptor haplotype to risk of death and heart transplantation in patients with heart failure. Am J Cardiol 2007; 99: 250-255.
- Savva, J., Maqbool A., White HL., Galloway SL., Yuldasheva NY., et al. Polymorphisms of adrenoceptors are not associated with an increased risk of adverse event in heart failure: a MERIT-HF substudy. J Card Fail 2009; 15: 435-44.
- Liggett, SB., et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med 2008; 14: 510-517.
- Wang, WC., et al. A polymorphism of G-protein coupled receptor kinase5 alters agonist-promoted desensitization of beta2-adrenergic receptors. Pharmacogenet Genomics 2008; 18: 729-732.
- Cresci, S., Kelly, R.J., Cappola, T.P., Diwan, A., Dries, D., et al. Clinical and genetic modifiers of long-term survival in heart failure. J Am Coll Cardiol 2009; 54: 432-444.
- Dorn, G.W. Genetics of common forms of heart failure. Curr Opin Cardiol 2011; 26: 204-208.
- Winker, R., Barth, A., Valic, E., Maier, R., Osterode, W., et al. Functional adrenergic receptor polymorphisms and idiopathic orthostatic intolerance. Int Arch Occup Environ Health 2005; 78: 171-177.
- Cho, S., Kim, C.H., Cubells, J.F., Zabetian, C.P., Hwang, D.Y., et al. Variations in the dopamine beta-hydroxylase gene are not associated with the autonomic disorders, pure autonomic failure, or multiple system atrophy. Am J Med Genet A 2003; 120A: 234-236.
- Timberlake, DS., O'Connor, DT., Parmer, RJ. Molecular genetics of essential hypertension: recent results and emerging strategies. Curr Opin Nephrol Hypertens 2001; 10: 71-79.
- Krushkal, J., Xiong, M., Ferrell, R., Sing, CF., Turner, ST., et al. Linkage and association of adrenergic and dopamine receptor genes in the distal portion of the long arm of chromosome 5 with systolic blood pressure variation. Hum Mol Genet 1998; 7: 1379-1383.
- Krushkal, J., Ferrell, R., Mockrin, SC., Turner, ST., Sing, CF., et al. Genome-wide linkage analyses of systolic blood pressure using highly discordant siblings. Circulation 1999; 99: 1407-1410.
- Bray, M.S., Krushkal J., Li, L., FerrelL, R., Kardia, S., et al. Positional genomic analysis identifies the beta(2)-adrenergic receptor gene as a susceptibility locus for human hypertension. Circulation 2000; 101: 2877-2882.
- Busjahn, A., Li, G.H., Faulhaber, H.D., Rosenthal, M., Becker, A., et al. beta-2 adrenergic receptor gene variations, blood pressure, and heart size in normal twins. Hypertension 2000; 35: 555-560.
- Hoit, B. D. et al., 2000. Beta2-adrenergic receptor polymorphisms at amino acid 16 differentially influence agonist-stimulated blood pressure and peripheral blood flow in normal individuals. Am Heart J, 139, 537-542.
- Wu, H., Tang, W., Li, H., Zhou, X., Yang, Y., et al. Association of the beta2-adrenergic receptor gene with essential hypertension in the non-Han Chinese Yi minority human population. J Hypertens 2006; 24: 1041-1047.
- Freitas, S.R., Pereira, A.C., Floriano, M.S., Mill, J.G., Krieger, J.E. Association of alpha1a-adrenergic receptor polymorphism and blood pressure phenotypes in the Brazilian population. BMC Cardiovasc Disord 2008; 8: 40.
- Xie, HG., Kim, RB., Stein, CM., Gainer, JV., Brown NJ., et al. Alpha1A-adrenergic receptor polymorphism: association with ethnicity but not essential hypertension. Pharmacogenetics 1999; 9: 651-656.
- Gu, D., Ge, D., Snieder, H., He, J., Chen, S., et al. Association of alpha1A adrenergic receptor gene variants on chromosome 8p21 with human stage 2 hypertension. J Hypertens 2006; 24: 1049-1056.
- Daly, R.N., Goldberg, P.B., Roberts, J.Effects of age on neurotransmission at the cardiac sympathetic neuroeffector junction. J Pharmacol Exp Ther 1988; 245: 798-803.
- Snyder, DL., Aloyo, VJ., Wang, W., Roberts, J. Influence of age and dietary restriction on norepinephrine uptake into cardiac synaptosomes. J Cardiovasc Pharmacol 1998; 32: 896-90.
- Rumantir, MS., Kaye, DM., Jennings, GL., Vaz, M., Hastings, JA., et al. Phenotypic evidence of faulty neuronal norepinephrine reuptake in essential hypertension. Hypertension 2000; 36: 824-829.
- Chitbangonsyn, S.W., Mahboubi, P., Walker, D., Rana, B.K., Diggle, K.L., et al. Physical mapping of autonomic/sympathetic candidate genetic loci for hypertension in the human genome: a somatic cell radiation hybrid library approach. J Hum Hypertens 2003; 17: 319-324.
- Esler, M., Eikelis, N., Schlaich, M., Lambert. G., Alvarenga, M., et al. Human sympathetic nerve biology: parallel influences of stress and epigenetics in essential hypertension and panic disorder. Ann N Y Acad Sci 2008; 1148: 338-348.
- Ono, K., Iwanaga, Y., Mannami, T., Kokubo, Y., Tomoike, H., et al. Epidemiological evidence of an association between SLC6A2 gene polymorphism and hypertension. Hypertens Res 2003; 26: 685-689.
- Zolk O., Ott C., Fromm MF., Schmieder, RE. Effect of the rs168924 single-nucleotide polymorphism in the SLC6A2 catecholamine transporter gene on blood pressure in Caucasians. J Clin Hypertens (Greenwich) 2012; 14: 293-298.
- Eisenhofer, G. The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol Ther 2001; 91: 35-62.
- Jia, H., Hingorani, A.D., Sharma, P., Hopper, R., Dickerson, C., et al. Association of the G(s)alpha gene with essential hypertension and response to beta-blockade. Hypertension 1999; 34: 8-14.
- Cabadak, H., Orun, O., Nacar, C., Dogan, Y., Guneysel, O., et al. The role of G protein β3 subunit polymorphisms C825T, C1429T, and G5177A in Turkish subjects with essential hypertension. Clin Exp Hypertens 2011; 33: 202-208.
- Niu, W., Qi, Y. Association of α-adducin and G-protein β3 genetic polymorphisms with hypertension: a meta-analysis of Chinese populations. PLoS One 2011; 6: e17052.
- Filigheddu, F., Argiolas, G., Degortes, S., Zaninello, R., Frau F., et al. Haplotypes of the adrenergic system predict the blood pressure response to beta-blockers in women with essential hypertension. Pharmacogenomics 2010; 11: 319-325.
- Beige, J., Hohenbleicher, H., Distler, A., Sharma, A.M. G-Protein beta3 subunit C825T variant and ambulatory blood pressure in essential hypertension. Hypertension 1999; 33: 1049-105.
- Sharma, P., Hingorani, A., Jia, H., Ashby, M., Hopper, R., et al. Positive association of tyrosine hydroxylase microsatellite marker to essential hypertension. Hypertension 1998; 32: 676-682.
- Nielsen, SJ., Jeppesen, J., Torp-Pedersen, C., Hansen, TW., Linneberg, A., et al. Tyrosine hydroxylase polymorphism (C-824T) and hypertension: a population-based study. Am J Hypertens 2010; 23: 1306-131.
- Abe, M., Wu, Z., Yamamoto M., Jin JJ., Tabara, Y., et al. Association of dopamine beta-hydroxylase polymorphism with hypertension through interaction with fasting plasma glucose in Japanese. Hypertens Res 2005; 28: 215-22.
- Chen, Y., Zhang, K., Wen, G., Rao, F., Sanchez, A.P., et al. Human dopamine β-hydroxylase promoter variant alters transcription in chromaffin cells, enzyme secretion, and blood pressure. Am J Hypertens 2011; 24: 24-32.
- Jacob, H.J., Lindpaintner, K., Lincoln, S.E., Kusumi, K., Bunke,r RK., et al. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell 1991; 67: 213-224.
- Cui, J., Zhou, X., Chazaro, I., DeStefano, AL., Manolis, AJ., et al. Association of polymorphisms in the promoter region of the PNMT gene with essential hypertension in African Americans but not in whites. Am J Hypertens 2003; 16: 859-863.
- Huang, C., Zhang, S., Hu, K., Ma, Q., Yang, T. Phenylethanolamine N-methyltransferase gene promoter haplotypes and risk of essential hypertension. Am J Hypertens 2011; 24: 1222-1226.
- Fava, C., Montagnana, M., Danese, E., Sjögren, M., Almgren, P., et al. The Renalase Asp37Glu polymorphism is not associated with hypertension and cardiovascular events in an urban-based prospective cohort: the Malmö Diet and cancer study. BMC Med Genet 2012; 13: 57.
- Buraczynska, M., Zukowski, P., Buraczynska, K., Mozul, S., Ksiazek, A. Renalase gene polymorphisms in patients with type 2 diabetes, hypertension and stroke. Neuromolecular Med 2011; 13: 321-327.
- Stec, A., Semczuk, A., Furmaga, J., Ksiazek, A., Buraczynska, M. Polymorphism of the renalase gene in end-stage renal disease patients affected by hypertension. Nephrol Dial Transplant 2012; 27: 4162-4166.
- Kamide, K., Kokub,o Y., Yang, J., Matayoshi, T., Inamoto, N., et al. Association of genetic polymorphisms of ACADSB and COMT with human hypertension. J Hypertens 2007; 25: 103-110.
- Hagen, K., Pettersen, E., Stovner, L.J., Skorpen, F., Holmen, J., et al. High systolic blood pressure is associated with Val/Val genotype in the catechol-o-methyltransferase gene. The Nord-Trøndelag Health Study (HUNT). Am J Hypertens 2007; 20: 21-26.
- Annerbrink, K., Westberg, L., Nilsson, S., Rosmond, R., Holm, G., et al. Catechol O-methyltransferase val158-met polymorphism is associated with abdominal obesity and blood pressure in men. Metabolism 2008; 57: 708-71.
- Htun, N.C., Miyaki, K., Song, Y., Ikeda, S., Shimbo, T., et al. Association of the catechol-O-methyl transferase gene Val158Met polymorphism with blood pressure and prevalence of hypertension: interaction with dietary energy intake. Am J Hypertens 2011; 24: 1022-1026.
- Zhang, WS., Xu, L., Schooling, CM., Jiang, CQ., Cheng, KK., et al. Effect of alcohol and aldehyde dehydrogenase gene polymorphisms on alcohol-associated hypertension: the Guangzhou Biobank Cohort Study. Hypertens Res 2013; 36: 741-746.
- Wei, Z., Biswas, N., Wang, L., Courel, M., Zhang, K., et al. A common genetic variant in the 3'-UTR of vacuolar H+-ATPase ATP6V0A1 creates a micro-RNA motif to alter chromogranin A processing and hypertension risk. Circ Cardiovasc Genet 2011; 4: 381-389.
- Wu, T., Snieder, H., de Geus, E. Genetic influences on cardiovascular stress reactivity. Neurosci Biobehav Rev 2010; 35: 58-68.
- Turpeinen, AK., Vanninen, E., Magga, J., Tuomainen, P., Kuusisto J., et al. Cardiac sympathetic activity is associated with inflammation and neurohumoral activation in patients with idiopathic dilated cardiomyopathy. Clin Physiol Funct Imaging 2009; 29: 414-419.
- Sharkey, SW., Maron, BJ., Nelson, P., Parpart, M., Maron, MS., et al. Adrenergic receptor polymorphisms in patients with stress (tako-tsubo) cardiomyopathy. J Cardiol 2009; 53: 53-57.
- Regitz-Zagrosek, V., Hocher, B., Bettmann, M., Brede, M., Hadamek, K., et al. Alpha2C-adrenoceptor polymorphism is associated with improved event-free survival in patients with dilated cardiomyopathy. Eur Heart J 2006; 27: 454-459.
- Forleo, C., Resta, N., Sorrentino, S., Guida, P., Manghisi, A., et al. Association of beta-adrenergic receptor polymorphisms and progression to heart failure in patients with idiopathic dilated cardiomyopathy. Am J Med 2004; 117: 451-458.
- Spinelli, L., Trimarco, V., Di Marino, S., Marino, M., Iaccarino, G., et al. L41Q polymorphism of the G protein coupled receptor kinase 5 is associated with left ventricular apical ballooning syndrome. Eur J Heart Fail 2010; 12: 13-16.
- Vriz, O., Minisini, R., Citro, R., Guerra, V., Zito C., et al. Analysis of beta1 and beta2-adrenergic receptors polymorphism in patients with apical ballooning cardiomyopathy. Acta Cardiol 2011; 66: 787-790.
- Figtree, G.A., Bagnall, R.D., Abdulla, I., Buchholz, S., Galougahi, K.K., et al. No association of G-protein-coupled receptor kinase 5 or β-adrenergic receptor polymorphisms with Takotsubo cardiomyopathy in a large Australian cohort. Eur J Heart Fail 2013; 15: 730-733.
- Norcliffe-Kaufmann, L., Axelrod, F., Kaufmann, H. Afferent baroreflex failure in familial dysautonomia. Neurology 2010; 75: 1904-191.
- Goldstein D.S., Holmes, C., Axelrod. F.B. Plasma catechols in familial dysautonomia: a long-term follow-up study. Neurochem Res 2008; 33: 1889-1893.
- Jain, S., Goldstein, D.S. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol Dis 2012; 46: 572-580.
- Kariya, S., Hirano, M., Takahashi, N., Furiya, Y., Ueno, S. Lack of association between polymorphic microsatellites of the VMAT2 gene and Parkinson's disease in Japan. J Neurol Sci 2005; 232: 91-94.
- García-Gorostiaga, I., Sierra, M., Sánchez-Juan, P., Ruiz-Martínez, J., Gorostidi, A., et al. Genetic variation in α-synuclein kinases (CK-2β and GRK-5) and risk of Parkinson's disease. Parkinsonism Relat Disord 2011; 17: 496-497.
- Costa, A., Riedel, M., Müller, U., Möller, H.J., Ettinger, U. Relationship between SLC6A3 genotype and striatal dopamine transporter availability: a meta-analysis of human single photon emission computed tomography studies. Synapse 2011; 65: 998-1005.
- Freeman, R., et al., 201. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res, 21, 69-72.
- Nickander, KK., Carlson, PJ., Urrutia, RA., Camilleri, M., Low, PA. A screen of candidate genes and influence of beta2-adrenergic receptor genotypes in postural tachycardia syndrome. Auton Neurosci 2005; 120: 97-103.
- Garland, E.M., Black, B.K., Harris, P.A., Robertson, D. Dopamine-beta-hydroxylase in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol 2007; 293: H684-H690.
- Nakao, R., Tanaka, H., Takitani, K., Kajiura, M., Okamoto, N., et al. GNB3 C825T polymorphism is associated with postural tachycardia syndrome in children. Pediatr Int 2012; 54: 829-837.
- Priori, SG., Napolitano, C., Memmi, M., Colombi, B., Drago, F., et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002; 106: 69-74.
- Mosqueda-Garcia, R., Furlan, R., Tank, J., Fernandez-Violante, R. The elusive pathophysiology of neurally mediated syncope. Circulation 2000; 102: 2898-2906.
- Marquez, MF., Hernandez-Pacheco, G., Hermosillo, AG., Gomez, JR., Cardenas, M., et al. The Arg389Gly beta1-adrenergic receptor gene polymorphism and susceptibility to faint during head-up tilt test. Europace 2007; 9: 585-588.
- Alboni, P., Brignole M., Degli, Uberti, E.C. Is vasovagal syncope a disease? Europace 2007; 9: 83-87.
- Pitzalis, M., Parati, G., Massari, F., Guida, P., Di Rienzo, M., et al. Enhanced reflex response to baroreceptor deactivation in subjects with tilt-induced syncope. J Am Coll Cardiol 2003; 41: 1167-1173.
- Vaddadi, G., Esler, MD., Dawood, T. Lambert E Persistence of muscle sympathetic nerve activity during vasovagal syncope. Eur Heart J 2010; 31: 2027-2033.
- Grass,i G. Vasovagal syncope, sympathetic mechanisms and prognosis: the shape of things to come. Eur Heart J 2010; 31: 1951-1953.
- Duplyakov. D., Golovina. G., Sysuenkova. E., Garkina. S. Can the result of a tilt test be predicted in the first five minutes? Cardiol J 2011; 18: 521-526.
- Porges, SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol 2001; 42: 123-146.
- Olde Nordkamp, LR., Wieling, W., Zwinderman, AH., Wilde, AA., van Dijk, N. Genetic aspects of vasovagal syncope: a systematic review of current evidence. Europace 2009; 11: 414-420.
- Cooper, C.J., Ridker, P., Shea, J., Creager, MA. Familial occurrence of neurocardiogenic syncope. N Engl J Med 1994; 331: 205.
- Mathias, CJ., Deguchi, K., Bleasdale-Barr, K., Kimber, JR. Frequency of family history in vasovagal syncope. Lancet 1998; 352: 33-34.
- Mathias, CJ., Deguchi, K., Bleasdale-Barr, K., Smith, S. Familial vasovagal syncope and pseudosyncope: observations in a case with both natural and adopted siblings. Clin Auton Res 2000; 10: 43-45.
- Newton, JL., Donaldson, P., Parry, S., Kenny, RA., Smith, J., et al. Angiotensin converting enzyme insertion/deletion polymorphisms in vasovagal syncope. Europace 2005; 7: 396-399.
- Sorrentino, S., et al. Endothelin system polymorphisms in tilt test-induced vasovagal syncope. Clin Auton Res 2009; 19: 123-129.
- La Rosee, K., Huntgeburth, M., Rosenkranz, S., Bohm, M., Schnabel, P. The Arg389Gly beta1-adrenoceptor gene polymorphism determines contractile response to catecholamines. Pharmacogenetics 2004; 14: 711-716.
- Hernández-Pacheco, G., Serrano, H., Márquez, M.F., Hermosillo, AG., Pérez-Vielma, N., et al. Genetic study of the vasovagal syncope associated to the Arg389Gly polymorphism of the beta1 adrenergic receptor. Arch Cardiol Mex 2008; 78: 134-138.
- Sorrentino, S., Forleo, C., Iacoviello, M., Guida, P., D'Andria V., et al. Lack of association between genetic polymorphisms affecting sympathetic activity and tilt-induced vasovagal syncope. Auton Neurosci 2010; 155: 98-103.
- Piccardi, M., Congiu, D., Squassina, A., Manconi, F., Putzu, PF., et al. Alzheimer's disease: case-control association study of polymorphisms in ACHE, CHAT, and BCHE genes in a Sardinian sample. Am J Med Genet B Neuropsychiatr Genet 2007; 144B: 895-899.
- Shi, J., Hattori, E., Zou, H., Badner, JA., Christian, SL., et al. No evidence for association between 19 cholinergic genes and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 2007; 144B: 715-723.
- Grunblatt, E., Reif, A., Jungwirth, S., Galimberti, D., Weber, H., et al. Genetic variation in the choline O-acetyltransferase gene in depression and Alzheimer's disease: the VITA and Milano studies. J Psychiatr Res 2011; 45: 1250-1256.
- Mancama, D., Mata, I., Kerwin, RW., Arranz, MJ. Choline acetyltransferase variants and their influence in schizophrenia and olanzapine response. Am J Med Genet B Neuropsychiatr Genet 2007; 144B: 849-853.
- Livolsi, A., Feldman, J., Feingold, J., Weiss, L., Alembik, Y., et al. First model of spontaneous vagal hyperreactivity and its mode of genetic transmission. Circulation 2002; 106: 2301-2304.
- Lara, A., Damasceno, DD., Pires, R., Gros, R., Gomes, ER., et al. Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol Cell Biol 2010; 30: 1746-1756.
- Prado, VF., Martins-Silva, C., de Castro, BM., Lima, RF., Barros, DM., et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 2006; 51: 601-612.
- Zoccoli, G., et al. Regulation of cerebral circulation during sleep. In Parmeggiani, P. L. and Velluti, R. (Eds.) The Physiological Nature of Sleep. 2005 (1st edtn). Imperial College Press: London.
- Bojic, T.Mechanisms of cardiovascular control and effects of acoustic stimulation on cardiovascular system during the wake-sleep cycle.2003.
- Floras, JS. Clinical aspects of sympathetic activation and parasympathetic withdrawal in heart failure. J Am Coll Cardiol 1993; 22: 72A-84A.
- Ishise, H., Asanoi, H., Ishizaka, S., Joho, S., Kameyama, T., et al. Time course of sympathovagal imbalance and left ventricular dysfunction in conscious dogs with heart failure. J Appl Physiol (1985) 1998; 84: 1234-124.
- Svingos, AL., et al. Vesicular acetylcholine transporter in the rat nucleus accumbens shell: subcellular distribution and association with mu-opioid receptors. Synapse 2001; 40: 184-192.
- Martins-Silva, C. et al. Novel strains of mice deficient for the vesicular acetylcholine transporter: insights on transcriptional regulation and control of locomotor behavior. PLoS One 2001; 6: e1761.
- Schmid, S., Azzopardi, E., De Jaeger, X., Prado, MA., Prado, VF. VAChT knock-down mice show normal prepulse inhibition but disrupted long-term habituation. Genes Brain Behav 2011; 10: 457-464.
- Tayebati, SK., et al. Vesicular acetylcholine transporter (VAChT) in the brain of spontaneously hypertensive rats (SHR): effect of treatment with an acetylcholinesterase inhibitor. Clin Exp Hypertens 2008; 30: 732-43.
- Lurie, KG., Dutton, J., Mangat, R., Newman, D., Eisenberg, S., et al. Evaluation of edrophonium as a provocative agent for vasovagal syncope during head-up tilt-table testing. Am J Cardiol 1993; 72: 1286-1290.
- Smith, R., Chung, H., Rundquist, S., Maat-Schieman, ML., Colgan, L., et al. Cholinergic neuronal defect without cell loss in Huntington's disease. Hum Mol Genet 2006; 15: 3119-313.
- Sheldon, RS., Wright, CI., Duff, HJ., Thakore, E., Gillis, AM., et al. Mechanism of hypotensive transients associated with abrupt bradycardias in conscious rabbits. Can J Cardiol 2007; 23: 721-726.
- Medina, A., Bodick, N., Goldberger, AL., Mac Mahon, M., Lipsitz, LA. Effects of central muscarinic-1 receptor stimulation on blood pressure regulation. Hypertension 1997; 29: 828-834.
- Friederich, H.C., Michaelsen, J., Hesse, C., Schellberg, D., Schwab, M., et al. Treatment of recurrent neurocardiogenic syncope with cardiac inhibitors with ipratropium bromide. Z Kardiol 2004; 93: 479-485.
- Livolsi, A. et al. Constitutive overexpression of muscarinic receptors leads to vagal hyperreactivity. PLoS One 2010; 5: e15618.
- Lelonek, M., Pietrucha, T., Stanczyk, A., Goch, JH. Vasovagal syncope patients and the C825T GNB3 polymorphism. Anadolu Kardiyol Derg 2007; 7 Suppl 1: 206-208.
- Lelonek, M. et al. A novel approach to syncopal patients: association analysis of polymorphisms in G-protein genes and tilt outcome. Europace, 2009; 11: 89-93.
- Lelonek, M., Zelazowska, M., Pietrucha, T. Genetic variation in protein as a new indicator in screening test for vasovagal syncope. Circ J 2011; 75: 2182-2186.
- Mitro, P., Rybarova, E., Zemberova, E., Tkac, I. Enhanced plasma catecholamine and cAMP response during the head-up tilt test in patients with vasovagal syncope. Wien Klin Wochenschr 2005; 117: 353-358.
- Suzuki, N. Roles of cerebrovascular innervations and their neurotransmitters in various pathological conditions of central nervous system. Rinsho Shinkeigaku 2012; 52: 819-824.
- Brignole, M., Alboni, P., Benditt, D., Bergfeldt, L., Blanc, J,J., et al. Guidelines on management (diagnosis and treatment) of syncope. Eur Heart J 2001; 22: 1256-1306.
- Parry, SW., Nath, S., Bourke, JP., Bexton, RS., Kenny, RA. Adenosine test in the diagnosis of unexplained syncope: marker of conducting tissue disease or neurally mediated syncope? Eur Heart J 2006; 27: 1396-1400.
- Shen, WK., Kurachi, Y. Mechanisms of adenosine-mediated actions on cellular and clinical cardiac electrophysiology. Mayo Clin Proc 1995; 70: 274-29.
- Moss, AJ. Clinical management of patients with the long QT syndrome: drugs, devices, and gene-specific therapy. Pacing Clin Electrophysiol 1997; 20: 2058-2060.
- Henrion, U., Zumhagen, S., Steinke, K., Strutz-Seebohm, N., Stallmeyer, B., et al. Overlapping cardiac phenotype associated with a familial mutation in the voltage sensor of the KCNQ1 channel. Cell Physiol Biochem 2012; 29: 809-818.
- Ernst, P.B., Garrison, J.C., Thompson, L.F .Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol 2010; 185: 1993-1998.
- Liu, X., Wang, F., Knight, AC., Zhao, J., Xiao, J. Common variants for atrial fibrillation: results from genome-wide association studies. Hum Genet 2012; 131: 33-39.
- Chen, Y.H., Xu, S.J., Bendahhou, S., Wang, X.L., Wang, Y., et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003; 299: 251-254.
- Brugada, R., Tapscott, T., Czernuszewicz, GZ., Marian, AJ., Iglesias, A., et al. Identification of a genetic locus for familial atrial fibrillation. N Engl J Med 1997; 336: 905-91.
- Ellinor, P.T., Shin, J.T., Moore, R.K., Yoerger, D.M., MacRae, C.A. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation 2003; 107: 2880-2883.
- Wakili, R., Voigt, N., Kaab, S., Dobrev, D., Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 2011; 121: 2955-2968.
- Mahida, S., Lubitz, SA., Rienstra, M., Milan, DJ., Ellinor, PT. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc Res 2011; 89: 692-700.
- Nattel, S. Atrial electrophysiology and mechanisms of atrial fibrillation. J Cardiovasc Pharmacol Ther 2003; 8 Suppl 1: S5-1.
- Huang, J.L., Wen, Z.C., Lee, W.L., Chang, M.S., Chen S.A. Changes of autonomic tone before the onset of paroxysmal atrial fibrillation. Int J Cardiol 1998; 66: 275-283.
- Herweg, B., Dalal, P., Nagy, B., Schweitzer, P. Power spectral analysis of heart period variability of preceding sinus rhythm before initiation of paroxysmal atrial fibrillation. Am J Cardiol 1998; 82: 869-874.
- Fuster, V., et al. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation, 2006; 114:e257-e354.
- Bettoni, M. Zimmermann, M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 2002; 105: 2753-2759.
- Patterson, E., Lazzara, R., Szabo, B., Liu, H., Tang, D., et al. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol 2006; 47: 1196-1206.
- Amar, D., Zhang, H., Miodownik, S., Kadish, A.H. Competing autonomic mechanisms precede the onset of postoperative atrial fibrillation. J Am Coll Cardiol 2003; 42: 1262-1268.
- Chou, C.C., Chen, P.S. New concepts in atrial fibrillation: neural mechanisms and calcium dynamics. Cardiol Clin 2009; 27: 35-43.
- Ohno, K., Tsujino, A., Brengman, JM., Harper, CM., Bajzer, Z., et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA 2001; 98: 2017-2022.
- Brandon, E.P., Mellott, T., Pizzo, D.P., Coufal, N., D'Amour, K.A., et al. Choline transporter 1 maintains cholinergic function in choline acetyltransferase haploinsufficiency. J Neurosci 2004; 24: 5459-5466.
- Mori, K., Hara, Y., Saito, T., Masuda, Y., Nakaya, H. Anticholinergic effects of class III antiarrhythmic drugs in guinea pig atrial cells. Different molecular mechanisms.Circulation 1995; 91: 2834-2843.
- Zhao, QY., Huang, CX., Liang, JJ., Chen, H., Yang B., et al. Effect of vagal stimulation and differential densities of M2 receptor and IK,ACh in canine atria. Int J Cardiol 2008; 126: 352-358.
- Yang, YH., Zheng, QS., Li, J., Shang, FJ., Liu, T., et al. Age-related changes in the atrial muscarinic type 2 receptor and their effects on atrial fibrillation vulnerability in rabbits. Exp Gerontol 2009; 44: 572-578.
- Orr, WC., Stahl, ML., Whitsett, T., Langevin, E. Physiological sleep patterns and cardiac arrhythmias. Am Heart J 1979; 97: 128-129.
- Keyl C., Schneider A., Dambacher M., Bernardi L Time delay of vagally mediated cardiac baroreflex response varies with autonomic cardiovascular control. J Appl Physiol (1985) 2001; 91: 283-289.
- Kubista, H., Boehm, S. Molecular mechanisms underlying the modulation of exocytotic noradrenaline release via presynaptic receptors. Pharmacol Ther 2006; 112: 213-242.
- Kubista, H., Kosenburger, K., Mahlknecht, P., Drobny, H., Boehm, S. Inhibition of transmitter release from rat sympathetic neurons via presynaptic M(1) muscarinic acetylcholine receptors. Br J Pharmacol 2009; 156: 1342-1352.
- Stewart, A., Huang, J., Fisher, RA. RGS Proteins in Heart: Brakes on the Vagus. Front Physiol 2012; 3: 95.
- Siffert, W., Rosskopf, D., Siffert, G., Busch, S., Moritz, A., et al. Association of a human G-protein beta3 subunit variant with hypertension. Nat Genet 1998; 18: 45-48.
- Schreieck, J., Dostal, S., von Beckerath, N., Wacker, A., Flory, M., et al. C825T polymorphism of the G-protein beta3 subunit gene and atrial fibrillation: association of the TT genotype with a reduced risk for atrial fibrillation. Am Heart J 2004; 148: 545-550.
- Hexime,r S.P., Srinivasa, S.P., Bernstein, L.S., Bernard, J.L., Linder, M.E., et al. G protein selectivity is a determinant of RGS2 function. J Biol Chem 1999; 274: 34253-34259.
- Riddle, EL., Rana, BK., Murthy, KK., Rao, F., Eskin, E., et al. Polymorphisms and haplotypes of the regulator of G protein signaling-2 gene in normotensives and hypertensives. Hypertension 2006; 47: 415-420.
- Sartori, M., Ceolotto, G., Dorigatti, F., Mos, L., Santonastaso, M., et al. RGS2 C1114G polymorphism and body weight gain in hypertensive patients. Metabolism 2008; 57: 421-427.
- Lai, LP., et al. Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca(2+)-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban: an insight into the mechanism of atrial electrical remodeling. J Am Coll Cardiol 1999; 33: 1231-1237.
- Lai, LP., Su, MJ., Lin, JL., Lin, FY., Tsai, CH., et al. Changes in the mRNA levels of delayed rectifier potassium channels in human atrial fibrillation. Cardiology 1999; 92: 248-255.
- Yue, L., Melnyk, P., Gaspo, R., Wang, Z., Nattel, S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ Res 1999; 84: 776-784.
- Voigt, N., Li, N., Wang, Q., Wang, W., Trafford, AW., et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012; 125: 2059-2070.
- Cao, K., Xia, X., Shan, Q., Chen, Z., Chen, X., et al. Changes of sarcoplamic reticular Ca(2+)-ATPase and IP(3)-I receptor mRNA expression in patients with atrial fibrillation. Chin Med J (Engl) 2002; 115: 664-667.
- Bhuiyan, Z.A., Van den Berg, M.P., Van Tintelen, J.P., Bink-Boelkens, M.T., Wiesfeld AC., et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 2007; 116: 1569-1576.
- Nof, E., et al. Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm 2001; 8: 1546-1552.
- Shan, J., et al. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2012; 111: 708-717.
- Lai, LP., Su, MJ., Yeh, HM., Lin, JL., Chiang, FT., et al. Association of the human minK gene 38G allele with atrial fibrillation: evidence of possible genetic control on the pathogenesis of atrial fibrillation. Am Heart J 2002; 144: 485-490.
- Ehrlich, J.R., Zicha. S., Coutu. P., Hébert, T.E. Nattel S Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc Res 2005; 67: 520-528.
- Olson, TM., Alekseev, AE., Liu, XK., Park, S., Zingman, LV., et al. Kv.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet 2006; 15: 2185-219.
- Ellinor, P.T., Lunetta, K.L., Glazer, N.L., Pfeufer, A., Alonso, A., et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 2010; 42: 240-244.
- Fatini, C., Sticchi, E., Genuardi, M., Sofi, F., Gensini, F., et al. Analysis of minK and eNOS genes as candidate loci for predisposition to non-valvular atrial fibrillation. Eur Heart J 2006; 27: 1712-1718.
- Sotoodehnia, N., Siscovick, DS., Vatta, M., Psaty, BM., Tracy, RP., et al. Beta2-adrenergic receptor genetic variants and risk of sudden cardiac death. Circulation 2006; 113: 1842-1848.
- Zak, I., et al. 2008. Cigarette smoking, carrier state of A or G allele of 46A>G and 79C>G polymorphisms of beta2-adrenergic receptor gene, and the risk of coronary artery disease.Kardiol Pol, 66, 380-6; discussion 387.
- Frey, U. H., et al., 2009. A novel functional haplotype in the human GNAS gene alters Galphas expression, responsiveness to beta-adrenoceptor stimulation, and peri-operative cardiac performance. Eur Heart J, 30, 1402-1410.
- Mathias, CJ. Autonomic diseases: clinical features and laboratory evaluation. J Neurol Neurosurg Psychiatry 2003; 74 Suppl 3: iii31-4.
- Gaborit, N., et al., Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation 2005; 112: 471-478.
- Costantini, M., Crema, A. The electrocardiology of atrial fibrillation. Ital Heart J Suppl 2000; 1: 632-640.
- Rengo, G., Lymperopoulos, A., Leosco, D., Koch, WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol 2011; 50: 785-792.


