Keywords
Rotavirus; Prevalence; Genotype; Phylogenetic tree; Thailand
Introduction
Rotavirus group A (RVA) is one of the most common etiologies of severe diarrhea in infants and young children worldwide. It has been estimated that children under 5 years old are inflicted with gastroenteritis that results in 527,000 fatalities each year, with the highest rate in developing countries in Africa and Asia [1]. In Thailand, between 1997 and 2005, on average 42.6% of pediatric patients admitted to hospitals had severe dehydration due to diarrhea caused by rotavirus infection [2].
Rotavirus is classified as a member of genus Rotavirus in the Reoviridae family. These viruses are approximately 75 nm icosahedral non-enveloped particles, consisting of 11 segmented double stranded RNA genome enclosed in a triple-layered capsid. The RVA gene segments are monocistronic and encode for six structural (VP1-4, 6 and 7) and four non-structural proteins (NSP1- 4), except for the gene segment 11 which has two overlapping ORFs and codes for NSP5 and NSP6. Besides classification of rotavirus in A-G groups based on the antigenicity of the middle layer VP6, the two outer layer proteins VP7 and VP4 also provide the basis for the dual classification system into G and P genotypes, respectively [3]. To date 27 G-genotypes and 37 P-genotypes have been reported in humans and animals [4]. Globally, G1, G2, G3, G4, and G9 in combination with P[4], P[6], and P[8] are the most common rotavirus genotypes. Furthermore, some unusual genotypes of rotaviruses have been reported in several countries [5-7]. Thus the dual classification system is very useful to categorize the viruses in G/P combinations in which up to 120 genotypes could be found [8]. However, only a limited number of the combined G/P genotypes are common in humans, such as G1P[8], G2P[4], G3P[8], G4P[8] and G9P[8] with G1P[8], which are the prevalent types of human rotaviruses detected in most surveys. Other genotypes such as G5, G6, G8, G10, and G12 may also be co-circulating with P[4], P[8], P[9], and P[14] in various geographic regions of the world.
In Thailand, the fluctuation of predominant genotypes has also been detected since 1997 [9]. Furthermore, the uncommon human rotavirus strains such as G3P[3], G3P[9], G3P[10], G3P[19], and G12P[9] have been detected in some areas of Thailand [2,10]. Two rotavirus vaccines, Rotarix® (GlaxoSmithKlinne Biologicals), a monovalent G1P1A[8] human attenuated vaccine, and RotaTeq® (Merck&Co., Inc.), a pentavalent human bovine reassorted vaccine expressing human G1, G2, G3, G4, and P1A[8] have already been licensed in many countries. The efficacy of rotavirus vaccines vary among different populations. Following vaccination, the rates of mortality and hospitalization tend to reduce especially in developing countries [11,12] and even lower in populations of the developing countries with the highest burden of the disease [13]. These two licensed rotavirus vaccines were introduced in Thailand in 2006 and 2008, respectively, but are not included as a part of the national immunization program, thereby limiting their access for health care [14]. Because of great genetic diversity and interspecies transmission, the genetic reassortments of rotavirus strains are common. Thus, continuous rotavirus surveillance is important to monitor the new types or new G/P combinations to evaluate whether current vaccines cover the most prevalent genotypes circulating in Thailand.
In this study, we aimed to determine the distribution pattern of group A rotavirus genotypes and perform a phylogenetic analysis of rotaviruses circulating in infants and children with acute diarrhea who were hospitalized in Bamrasnaradura Infectious Diseases Institute, Department of Disease Control, Ministry of Public Health, Nonthaburi, Thailand.
Materials and Methods
Specimen collection and rotavirus detection
Children under age of five with moderate to severe diarrhea who were admitted to Bamrasnaradura Infectious Diseases Institute, Nonthaburi, Thailand, were screened for rotavirus infection by using a rapid qualitative immunochromatography assays (VIKIA Rota-Adeno, bioMerieux SA, France). 73 samples that were positive for rotavirus were collected anonymously from April 2012 to April 2014 and kept at -80°C for further examination. The study protocol was approved by the Institutional Ethical Committee of Bamrasnaradura Infectious Diseases Institute and Burapha University.
Viral RNA extraction
Fecal samples were diluted 10% in phosphate buffered saline (PBS) and clarified by centrifugation at 3500 g for 10 min. The supernatant fluid was collected and 140 μl of each was processed for viral RNA extraction with QIAamp® Viral RNA Mini Kit (QIAGEN, Germany).
Detection of group A rotavirus by RT-PCR
Partial-length VP7 (881 bp) and the VP8* region (876 bp) of VP4 genes were amplified separately by one step RT-PCR (SuperScript III one-step RT-PCR with Platinum Taq, USA). The reaction mixture consisted of 3 μl of extracted RNA, 12.5 μl of 2x reaction mix (Invitrogen, USA), SuperScriptIII RT/platinum Taq mix, 0.4 μM of each primer, 2.0 mM of MgSO4 in the total of 25 μl. A reaction mixture without a template was used as a negative control, and the reference strain, human rotavirus A strain Wa (ATCC 2018) RNA was used as a positive control. The VP7 gene was amplified with a primer pair VP7F and VP7R [15] to obtain a partial 881 bpgene fragment. The VP4 gene was amplified with a primer pair VP4 forward con3 and VP4 reverse con2 primer [16] to generate an 876 bp-partial gene fragment.
G and P genotyping by multiplex semi-nested PCR
The genotyping of group A rotaviruses was characterized by the multiplex semi-nested PCR reaction with a pool of internal type-specific forward primers and the reverse primer. One microliter of VP7 or VP4-gene product was used as the template. RNA of human rotavirus A strain Wa (ATCC 2018) and reaction without template were used as the positive and negative control, respectively.
To characterize G genotype, a mixture of forward primers, aBT1, aCT2, mG3, and mG9 in combination with the VP7 reverse primer [17] were utilized for amplification of the VP7 G1, G2, G3, and G9 gene fragment, respectively, in the PCR reaction. The reaction was carried out in a total of 25 μl consisting of 0.2 mM dNTPs mix, 2.0 mM MgCl2, 1.0 μM each of forward and reverse primer, 2.5 U Taq DNA polymerase (Thermoscientific, USA). Genotyping was based on size comparison of PCR products after direct visualization on 1.5% agarose gel. Expected DNA fragments of 618 (G1), 521 (G2), 682 (G3), and 179 (G9) bp were detected compared with 100 bp DNA Ladder marker (TrackIt, Invitrogen, USA).
For P genotyping, a mixture of 0.5 μM of each 2T-1, 3T-1, IT-1, and 4T-1 forward primers were used in combination with the VP4 reverse con2 primer [16] for amplification of the VP4 P[4], P[6], P[8], and P[9] gene fragment, respectively in the PCR reaction containing 0.2 mM dNTPs mix, 2.5 mM MgCl2, and 2.5 U Taq DNA polymerase (Thermoscientific, USA). Expected amplified products of 413 bp, 628 bp, 531 bp, and 502 bp DNA fragments represent P[4], P[6], P[8], and P[9] respectively visualized on 1.5% agarose gel.
Both G and P genotyping were performed under the same thermocycler condition as follows: initial denaturation at 95°C, 3 min; denaturation at 95°C, 30 sec; annealing at 55°C, 30 sec, extension at 72°C, 1 min in the total 30 cycles and followed by the final extension at 72°C, 10 min. The viruses with genotypes that could not be determined by multiplex PCR were subjected to identification by nucleotide sequencing.
Nucleotide sequencing and phylogenetic analysis
Partial-length cDNA were extracted from the agarose gel and purified with PureLink™ Quick Extraction kit (Invitrogen, USA). The nucleotide sequence of each DNA fragment of VP7 and VP4 gene was determined in both directions with the specific oligonucleotide sequencing primers; VP7-F primer or VP7-R primer for VP7 gene; VP4-F (con3) or VP4-R (con2) primers for VP4 gene (First Base Laboratories SDN BHD, Malaysia). The nucleotide sequences of the VP7 and VP4 genes were assembled and analyzed using BioEdit. The sequences were subsequently aligned and compared with those of the reference strains available in the NCBI GenBank database using BLAST search (www.ncbi.nlm.nih.gov/blast) and analyzed using CLUSTAL X program, version 1.83, for multiple sequence alignment, and MEGA4.1 for phylogenetic tree construction. Lineage designation was based on similarity to previously defined lineage reference strain. The statistical significance of the constructed phylogenies was analyzed using neighbor-joining method with the 1000 replicates. Branches with bootstrap number below 50% were considered insignificant and excluded.
Nucleotide sequence accession number
Prototype sequences used in the phylogenetic analyses were retrieved from the GenBank database based on the closest nucleotide sequence relatedness with the strains sequenced in the present study. Prototypes G and P sequences of most common genotypes, lineages, and sub-lineages in human were also retrieved from GenBank and included in the phylogenetic analysis.
The nucleotide sequences of group A rotavirus described in the present study have been deposited in the GenBank database with the access numbers as follows: VP7 G1(KU588886- KU588899, KU588901-KU588902, KU588908, KU588912- KU588915, KU588919, KU588927-KU588928, KU588932- KU588936, KU588939,KU588942-KU588956), VP7 G2(KU588878- KU588884), VP7 G3(KU588859- KU588861) VP7 G8(KU588862- KU588877), and VP7G9 (KU991795-KU991796); VP4P[4] (KU754055-KU754060), and VP4P[8] (KU754064-KU754078, KU754080- KU754084, KU754087- KU754093, KU754098- KU754102, KU754106- KU754107, KU754109- KU754112, KU754116- KU754126, KU754128- KU754132, KU754134- KU754135, KU754137-KU754145).
Results
Distribution of group A rotavirus strains
The G- and P-genotype of 73 strains of group A rotaviruses were identified by RT-multiplex nested-PCR and nucleotide sequencing. Five different G-genotypes, i.e., G1, G2, G3, G8, and G9 were detected during this study period (Table 1). Among the rotavirus strains detected, G1 is the most predominant genotype (61.64%; N=45/73), followed by G8 (21.92%; N=16/73), G2 (9.59%; N=7/73), G3 (4.11%; N=3/73), and G9 (2.74%; N=2/73). G4, a common genotype, was not found in any rotavirus positive samples by nucleotide sequence analysis of partial length VP7 gene. When examined for their P-genotypes, only P[4] and P[8] were detected. The majority of rotavirus isolates were P[8] 91.78% (N=67/73) whereas only a small proportion were P[4] 8.22%. (N=6/73). All of G1, G3, G8, and G9 rotavirus strains were found exclusively with P[8], while G2 strains were found exclusively with P[4]. However, only one G2 isolate coexisted with P[8]. The G- and P-genotype combinations shown in Table 1 indicate that G1P[8] was the predominant genotype (61.64%; 45/73), followed by G8P[8] (21.92%; 16/73), G2P[4] (8.22%; 6/73), G3P[8] (4.11%; 3/73), G9P[8] (2.74%; 2/73), G2P[8] (1.37%; 1/73).
| P genotypes |
G Genotypes |
|
| G1 |
G2 |
G3 |
G8 |
G9 |
Total |
| P[4] |
- |
6
(8.22%) |
- |
- |
- |
6
(8.22%) |
| P[8] |
45
(61.64%) |
1
(1.37%) |
3
(4.11%) |
16
(21.92%) |
2 (2.74%) |
67 (91.78%) |
| Total (%) |
45
(61.64%) |
7
(9.59%) |
3
(4.11%) |
16
(21.92%) |
2
(2.74%) |
73 (100%) |
Table 1: Distribution and relative frequencies of various combinations of G- and P-Genotypes GroupA rotavirus detected in hospitalized children with diarrhea.
Phylogenetic analysis of G-type rotaviruses
In order to gain insight into the degree of variability in the rotavirus strains currently circulating in Thailand, nucleotide sequences of rotaviruses obtained from 73 fecal samples were compared with the reference strains reported worldwide. Based on the phylogenetic analysis created for VP7 gene (Figure 1). G1 strains were classified into seven lineages (I-VII). Most G1 strains belonged to lineage II, sub-lineage IIc (Figure 1a). Those strains showed more than 93% similarity between themselves and their genes are closely related (94-99%) to strains previously identified in Japan, Australia, Cuba, the USA, Italy, India, Russia, and Thailand. G1 strains which were clustered into lineage I fell into sub-lineage Ic were segregated into 3 distinct branches, of which the major branch consisted of strains that shared 99% nucleotide similarity with other Thai strains previously reported (GQ996867CU163-KK/07 and GQ 996868 CU246-KK/08). The other three strains (KU588889Ash B-1014, KU588897Ash B-1034, and KU588893Ash B-1019) shared 96-97% identity with previously identified strains (DQ512969 VN-368 Vietnam, DQ207389 6193- 03 Ireland and AY098670 ISO-4 India),while another strain (KU588887 AshB-1011) in a separate branch shared 97-99% similarity with previously identified strains (AF183848 CH9 Taiwan, U26364 Ban-48 Bangladesh, GU985249 PJ13/07 China, GU979204 GER169-09 Germany, AB585918 8735/05 Hong Kong, and HM467952 LB2719 USA).


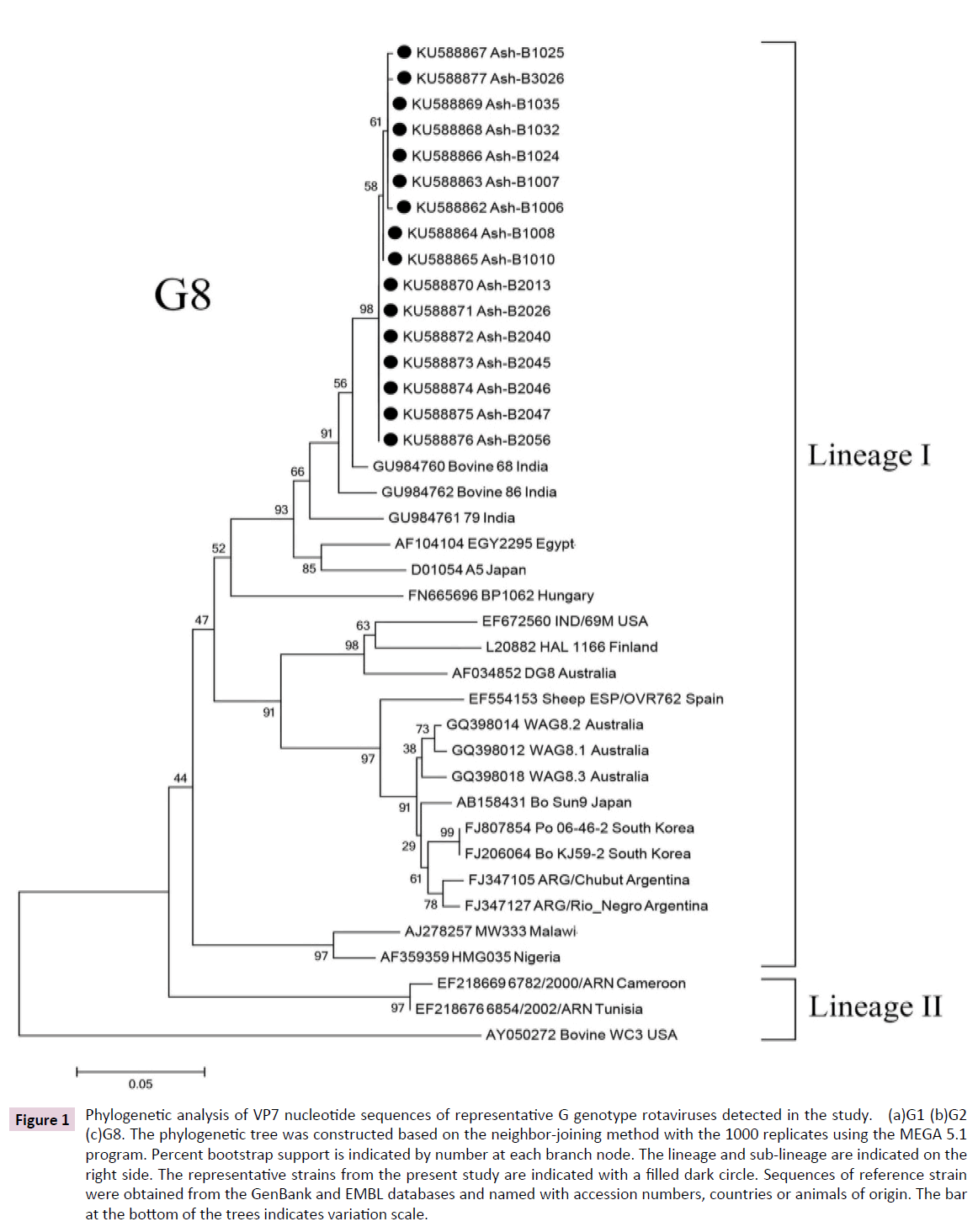
Figure 1: Phylogenetic analysis of VP7 nucleotide sequences of representative G genotype rotaviruses detected in the study. (a)G1 (b)G2 (c)G8. The phylogenetic tree was constructed based on the neighbor-joining method with the 1000 replicates using the MEGA 5.1 program. Percent bootstrap support is indicated by number at each branch node. The lineage and sub-lineage are indicated on the right side. The representative strains from the present study are indicated with a filled dark circle. Sequences of reference strain were obtained from the GenBank and EMBL databases and named with accession numbers, countries or animals of origin. The bar at the bottom of the trees indicates variation scale.
G2 could be classified into four lineages (I-IV). The VP7 nucleotide sequences of seven strains (KU588883 Ash-B1022, KU588884 Ash-B1027, KU588879 Ash-B1002, KU588881 Ash-B1020, KU588882 Ash-B1021, KU588878 Ash-B1001, and KU588880 Ash-B1012) of G2 belonged to lineage II, sub-lineage IIa and showed 100% similarity to each other (Figure 1b). All strains showed that their genetic backgrounds were closely related to the G2 strains previously identified in the USA, Australian, Uruguay, India, Kenya, Taiwan Japan, Vietnam, China, and Thailand from 2001 to 2003 [18] in the range of 96-99% similarity.
G3 could be classified into 2 lineages (I-II). All three strains of G3 (KU588859 Ash-B2022, KU588860 Ash-B2024, and KU588861 Ash-B2037) belonged to lineage I, sub-lineage Ia, and show 100% identity among themselves. These strains displayed more closely related genetic background to the G3 strains previously identified worldwide, such as in Thailand, China, Hong Kong, Vietnam, South Africa, Argentina, Italy, Germany, and the USA.
Interestingly, very rare G8 strains were also detected in this period of study at a relatively high frequency (21.92%). All 16 strains belonged to lineage I and showed VP7 nucleotide sequence with over 99 % similarity (Figure 1c). Their genetic backgrounds were more closely related to G8 bovine strains previously identified in India (96-98%).
In this period of study only two G9 strains (KU991795 and KU991796) were detected and were classified into lineage III with 99% nucleotide similarity among themselves and 91- 93% similarity with G9 strains previously identified in Belgium, Canada, Japan, China, India, and Thailand (AB436829 CHB058-02 Thailand and AB436825 MS057-02).
Phylogenetic analysis of P-types rotaviruses
Based on VP 4 gene, the total six strains of P[4] were detected in the present study (Figure 2a). All of these strains shared 100% sequence identity with each other, and were more closely related to strains detected in Myanmar with 99% similarity. Based on the analysis phylogenetic tree, all P[4] strains belonged to lineage V, sub-lineage Vb.
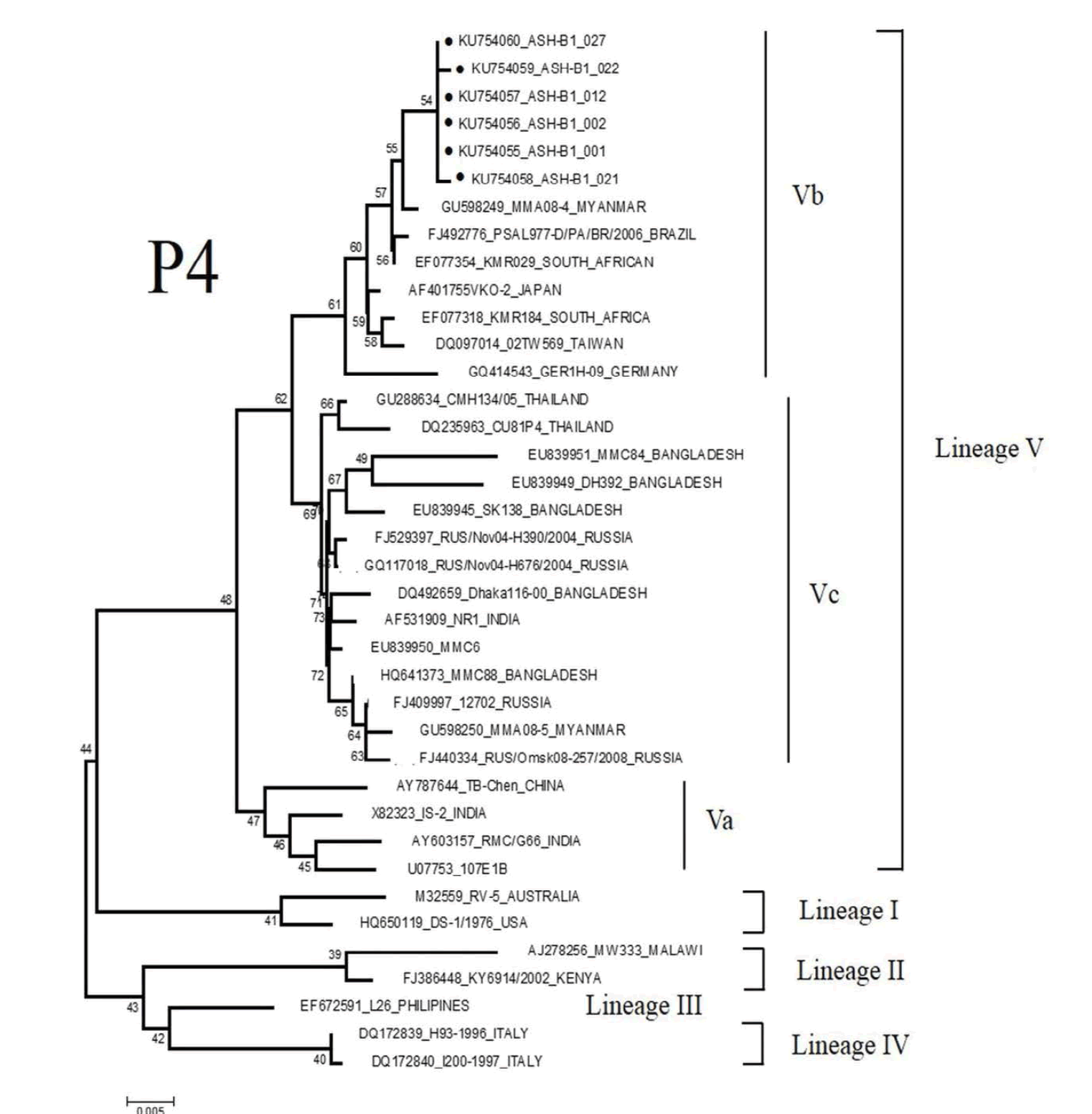

Figure 2: Phylogenetic analysis of VP4 nucleotide sequences of P genotype rotaviruses detected in the study: (a) P[4] (b) P[8]. The tree was constructed based on the neighbor-joining method with the 1000 replicates using the MEGA 5.1 program. Percent bootstrap support is indicated by a number at each branch node. The lineage and sub-lineage are indicated on the right side. The representative strains from the present study are indicated with a filled dark circle. Reference strain sequences were obtained from the GenBank and EMBL databases and named with accession numbers and countries or animals of origin. The bar at the bottom of the trees indicates variation scale.
A total of 67 P[8] strains identified in the present study belonged to lineage III and clustered in distinct branches (Figure 2b). These strains shared 96-100% nucleotide sequence identity to P[8] strains previously identified in Thailand.
Discussion
Rotavirus infection remains an important cause of mortality among young children in the developing countries [19]. Since natural changes in the prevalence of rotavirus strains are common, continuous monitoring of the circulating genotypes in the community is essential to ensure the delivery of efficacious interventions.
When compared to previous studies [20] from 1993 to 2011, the genotypes of rotavirus prevalence in Thailand have changed over time. The G1 genotype, a common strain worldwide [21], was still found with high frequency in the present study. In Thailand, G1 expressed a high incidence rate during 1993 to 1999, which subsequently decreased. From 2003 to 2004, the G1 genotype reached its peak and remains to be the predominant strain in Thailand. Sequence analysis of VP7 gene revealed that most of the G1 genotype detected belonged to the lineage II, sub-lineage IIc, and the lineage I, sub-lineage Ic which were closely related to the G1 genotype reported in other regions of Thailand (from 2004 to 2006 and 2007 to 2008) and the strains discovered worldwide. These findings indicated the genetic diversity of G1 and its wide distribution. In addition, G1 strains were observed exclusively in combination with P[8] that shared identical nucleotide sequence with P[8] strains previously identified in most parts of Thailand [22,23].
G2 has been found to be common worldwide. In Thailand the prevalence of G2 rotavirus has also changed over time. During 1993 to 2000 the prevalent rate of G2 in many regions of Thailand was much lower than those of G1[22, 23]. However, from 2002 to 2004, G2 became the dominant genotype in Bangkok [24] and co-dominant with G9 in other areas of the country [22]. The prevalence rate of G2 showed a fluctuating pattern from1999 to 2004 [22], then continuously decreased. During 2008 to 2009 the prevalence rate of G2 became co-predominant with G1 again [25] and then decreased to a very low frequency or was undetectable during 2009 to 2011 [20]. In the present study, G2P[4] was the third most predominant genotype after G1P[8] and G8P[8]. Based on the phylogenetic analysis, all strains belonged to lineage G2, sub-lineage IIa and seemed to be a single strain due to a 100% similarity to each other. A genetic distance analysis also showed that these G2 strains were closely related to G2 strains which were prevalent worldwide. This finding implies that G2 rotavirus has been prevailing and spreading throughout the world [18]. In addition, G2 have been detected exclusively in combination with P[4] genotype. Interestingly, only one case of G2 in combination with P[8] was detected in this study, which was also reported in a previous study in 2006 to 2007 [26]. It suggested that G2P[4] found in this surveillance might re-emerge from the original strains in the area. It is, therefore, important to monitor the trend and evolution of G2 in the next epidemic seasons.
G3 in Thailand have been reported at a far fewer frequency compared to G1 and G2 [10] or even undetectable in many areas of the country [22]. In the present study, the G3 genotype was detected at a low frequency which agreed with the long-term survey in Thailand. However, this trend appeared to be opposite to some areas of the country that showed a prevalence of the G3 genotype in KhonKaen and Bangkok [20]. The phylogenetic analysis revealed that the detected G3 strain belonged to lineage I, sub lineage Ia and is likely to be a single strain which showed a closer genetic relationship with G3 strains previously identified worldwide as well as in Bangkok, Thailand (DQ67493464vp7n) [2]. It suggested that G3P[8] found in this surveillance might have originated from the strains circulating in Bangkok, Thailand.
G9 was first reported in Bangkok at a very low frequency during 1994 to 1995 then disappeared afterwards [22]. During 2000 to 2002, it re-emerged and became the dominant genotype [22]. Subsequently, its prevalent pattern tended to fluctuate or was even undetectable until 2011 [20,25]. In the present study, G9 was found to be of lesser frequency than G3. However, in this study G9 strains from two positive samples were found in exclusive combination with P[8]. The strain was classified into lineage III whose members are spreading worldwide. Because of the close genetic relationship, the G9 rotavirus detected in the present study is likely derived from the G9 previously identified in Thailand [26]. In addition, the nucleotide sequence of their VP7 gene was also closely related to the strains detected in Belgium, Canada, Japan, China, and India, indicating that they might have descended from the same ancestor. Unlike other globally common G1, G2, G3, or G4 strains, which are detected almost exclusively with P[8] or P[4], the G9 strains have been detected in various P types [27].
A major finding in this study was that the globally uncommon genotype G8 in association with P[8] was detected at a relatively high frequency. It should be noted that the “Gouvea” primers showed cross-reactivity between types G3 and G8 [28] and DNA sequencing was necessary to confirm the VP7G8 genotyping results. The G8 rotavirus genotype is typical of strains infecting bovine and other animal host species, normally in combination with P[1], P[5], and P[21] types. In humans, the first G8 rotavirus was detected in an Indonesian child in 1980 [29], afterwards its identification in children with diarrhea has been reported sporadically worldwide especially in Africa [6], also in Sao Tome and Principe, Portugal, [30], and Italian regions [31]. In Thailand, G8 was first detected at a low percentage from 2003 to 2004 [22]. All G8 strains detected in the present study were closely related to bovine G8 strains previously identified in India (96- 98%) and sheep G8 strains previously identified in Spain (86%). According to the new RV-A classification system, the intragenotype nucleotide identity which utilizes a cutoff value of 80% for the VP7 gene [32], these two animal G8 strains effectively belong to the same genotype. It is possible that bovines were the main natural reservoir and the source of human rotavirus G8 strains identified in this study. Its appearance in human is likely to be the result of zoonotic transmission in the past. Interspecies transmission between humans and animals or animals and animals might have taken place in nature [33]. These interspecies and zoonotic transmissions and close proximity to humans with domestic animals could result in the reassortment of new rotavirus strains between human P[8] strains bearing G1, G3,G4, or G9 VP7 specificity and cocirculating bovine G8 strains bearing whether the P[4] or P[6] allele [30,34]. In this study the G8P[8] seems almost identical thus it probably excludes putative “epidemic outbreaks”.
However, analysis of the VP7 and VP4 genes may not be sufficient to provide a conclusive date on the possible origin of a rotavirus strain. The complete genome sequence information is necessary to obtain a better understating of the possibility of human-animal reassortments, cross-species transmission. It may also provide evidence of animal rotaviruses acting as reservoirs for one or several genes of human rotavirus strains. Since animal rotavirus or human-animal reassortants circulate in the population and in the environment, the monitoring of new emerging virus strains may help anticipate possible reduced efficacy of current vaccines to protect children against novel unconventional genotypes. Although both Rotarix™ and RotaTeq™ have shown high efficacy against the five major genotypes circulating worldwide and broad cross-protection against serotypes not included in the vaccine composition, efficacy data against genotypes of non-human origin remains limited [35].
Conclusion
G1P[8] genotype, the most common worldwide, was the most prevalent genotype in this study. The uncommon G8P[8] and G2P[8] genotypes were also identified. This present study has demonstrated the local fluctuation of genotype prevalence that frequently occurs over time, and emphasized the importance of continuous surveillance of rotavirus genotypes circulating in human and other animal species in order to obtain a better understanding of the epidemiology of rotavirus A infection within the region. More extensive epidemiological studies are also required to assess the potential effectiveness of current vaccines in our geographic location.
Acknowledgments
This work was supported by the office of the National Research Council of Thailand. We would like to thank Prof. Prasert Sobhon and Prof. Somsak Pantuwatana for advice, encouragement, and editing the manuscript.
20153
References
- Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, et al. (2012) 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. The Lancet Infectious Diseases 12: 136-141.
- Theamboonlers A, Bhattarakosol P, Chongsrisawat V, Sungkapalee T, Wutthirattanakowit N, et al. (2008) Molecular characterization of group A human rotaviruses in Bangkok and Buriram, Thailand during 2004-2006 reveals the predominance of G1P[8], G9P[8] and a rare G3P[19] strain. Virus Genes 36: 289-298.
- Knipe DM, Howley PM (2007) Fields' Virology: Lippincott Williams & Wilkins. J Gen Virol 1650.
- Trojnar E, Sachsenröder J, Twardziok S, Reetz J, Otto PH, et al. (2013) Identification of an avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J Gen Virol 94: 136-142.
- Agbemabiese CA, Nakagomi T, Doan YH, Nakagomi O (2015) Whole genomic constellation of the first human G8 rotavirus strain detected in Japan. Infect Genet Evol 35: 184-193.
- Gautam R, Mijatovic-Rustempasic S, Roy S, Esona MD, Lopez B, et al. (2015) Full genomic characterization and phylogenetic analysis of a zoonotic human G8P [14] rotavirus strain detected in a sample from Guatemala. Infect Genet Evol 33: 206-211.
- Saikruang W, Khamrin P, Chaimongkol N, Suantai B, Kongkaew A, et al. (2013) Genetic diversity and novel combinations of G4P[19] and G9P[19] porcine rotavirus strains in Thailand. Vet Microbiol 161: 255-262.
- Matthijnssens J, Bilcke J, Ciarlet M, Martella V, Bányai K, et al. (2009) Rotavirus disease and vaccination: impact on genotype diversity. Future Microbiol 4: 1303-1316.
- Khamrin P, Maneekarn N, Malasao R, Nguyen TA, Ishida S, et al. (2010) Genotypic linkages of VP4, VP6, VP7, NSP4, NSP5 genes of rotaviruses circulating among children with acute gastroenteritis in Thailand. Infect Genet Evol 10: 467-472.
- Khamrin P, Peerakome S, Tonusin S, Malasao R, Okitsu S, et al. (2007) Changing pattern of rotavirus G genotype distribution in Chiang Mai, Thailand from 2002 to 2004: decline of G9 and reemergence of G1 and G2. J Med Virol 79: 1775-1782.
- Armah GE, Sow SO, Breiman RF, Dallas MJ, Tapia MD, et al. (2010) Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in sub-Saharan Africa: a randomised, double-blind, placebo-controlled trial. Lancet 376: 606-614.
- Feikin DR, Laserson KF, Ojwando J, Nyambane G, Ssempijja V, et al. (2012) Efficacy of pentavalent rotavirus vaccine in a high HIV prevalence population in Kenya. Vaccine 30 Suppl 1: A52-A60.
- Organization WH (2010) Global vaccination update, 2009 = Le point sur la vaccination dans le monde, 2009. Weekly Epidemiological Record = Relevé épidémiologique hebdomadaire 85: 439-444.
- Muangchana C, Riewpaiboon A, Jiamsiri S, Thamapornpilas P, Warinsatian P (2012) Economic analysis for evidence-based policy-making on a national immunization program: a case of rotavirus vaccine in Thailand. Vaccine 30: 2839-2847.
- Iturriza-Gómara M, Kang G, Gray J (2004) Rotavirus genotyping: keeping up with an evolving population of human rotaviruses. J Clin Virol 31: 259-265.
- Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, et al. (1992) Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol 30: 1365-1373.
- Gouvea V, Glass RI, Woods P, Taniguchi K, Clark HF, et al. (1990) Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol 28: 276-282.
- Trinh QD, Pham NTK, Nguyen TA, Phan TG, Yan H, et al. (2010) Sequence analysis of the VP7 gene of human rotaviruses G2 and G4 isolated in Japan, China, Thailand, and Vietnam during 2001-2003. J Med Virol 82: 878-885.
- Morris SK, Awasthi S, Khera A, Bassani DG, Kang G, et al. (2012) Rotavirus mortality in India: estimates based on a nationally representative survey of diarrhoeal deaths. Bull World Health Organ 90: 720-727.
- Maiklang O VV, Vutithanachot C, Hacharoen P, Chieochansin T, Poovorawan Y(2012) Prevalence of group A genotype human rotavirus among children with dirarrhea in Thailand, 2009-2011. Southeast Asian J Trop Med Public Health 43: 904-916.
- Ruggeri FM, Bonomo P, Ianiro G, Battistone A, Delogu R, et al. (2015) Rotavirus Genotypes in Sewage Treatment Plants and in Children Hospitalized with Acute Diarrhea in Italy in 2010 and 2011. Appl Environ Microbiol 81: 241-249.
- Pongsuwannna Y, Guntapong R, Tacharoenmuang R, Prapanpoj M, Kameoka M, et al. (2010) A long-term survey on the distribution of the human rotavirus G type in Thailand. J Med Virol 82: 157-163.
- Chaimongkol N, Khamrin P, Malasao R, Thongprachum A, Ushijima H, et al. (2012) Genotypic linkages of gene segments of rotaviruses circulating in pediatric patients with acute gastroenteritis in Thailand. Infect Genet Evol 12: 1381-1391.
- Theamboonlers A, Veravigrom M, Yambangyang O, Trairatvorakul P, Chongsrisawat V, et al. (2005) The incidence of rotavirus a isolates of G genotype in Thailand in 2002-2004. Acta Virol 49: 111-115.
- Khananurak K, Vutithanachot V, Simakachorn N, Theamboonlers A, Chongsrisawat V, et al. (2010) Prevalence and phylogenetic analysis of rotavirus genotypes in Thailand between 2007 and 2009. Infect Genet Evol 10: 537-545.
- Kittigul L, Swangsri T, Pombubpa K, Howteerakul N, Diraphat P, et al. (2014) Rotavirus infection in children and adults with acute gastroenteritis in Thailand. Southeast Asian J Trop Med Public Health 45: 816-824.
- Hoshino Y, Jones RW, Ross J, Honma S, Santos N, et al. (2004) Rotavirus serotype G9 strains belonging to VP7 gene phylogenetic sequence lineage 1 may be more suitable for serotype G9 vaccine candidates than those belonging to lineage 2 or 3. J Virol 78: 7795-7802.
- Potgieter N, de Beer MC, Taylor MB, Steele AD (2010) Prevalence and diversity of rotavirus strains in children with acute diarrhea from rural communities in the Limpopo Province, South Africa, from 1998 to 2000. J Infect Dis 202 Suppl: S148-S155.
- Matsuno S, Hasegawa A, Mukoyama A, Inouye S (1985) A candidate for a new serotype of human rotavirus. J Virol 54: 623-624.
- Istrate C, Sharma S, Nordgren J, Videira e Castro S, Lopes Â, et al. (2015) High rate of detection of G8P[6] rotavirus in children with acute gastroenteritis in São Tomé and Príncipe. Arch Virol 160: 423-428.
- Ianiro G, Delogu R, Bonomo P, Castiglia P, Ruggeri FM, et al. (2014) Molecular characterization of human G8P[4] rotavirus strains in Italy: proposal of a more complete subclassification of the G8 genotype in three major lineages. Infect Genet Evol 21: 129-133.
- da Silva MFM, Tort LFL, Goméz MM, Assis RMS, Volotão EdM, et al. (2011) VP7 Gene of human rotavirus A genotype G5: Phylogenetic analysis reveals the existence of three different lineages worldwide. J Med Virol 83: 357-366.
- Cook N, Bridger J, Kendall K, Gomara MI, El-Attar L, et al. (2004) The zoonotic potential of rotavirus. J Infect 48: 289-302.
- Martella V, Bányai K, Matthijnssens J, Buonavoglia C, Ciarlet M (2010) Zoonotic aspects of rotaviruses. Vet Microbiol 140: 246-255.
- Yen C, Figueroa JR, Uribe ES, Carmen-Hernández LD, Tate JE, et al. (2011) Monovalent rotavirus vaccine provides protection against an emerging fully heterotypic G9P[4] rotavirus strain in Mexico. J Infect Dis 204: 783-786.