Keywords
Hyperthermia; Hippocampus; Kainite exytotoxicity; Hsp70; CA3; Heat stress
Introduction
As a default response to many injuries caused by hyperthermia, ischemia, excitotoxicity and a variety of pathological states including epilepsy, trauma, and neurodegenerative diseases, cells induce a variety of proteins, most notably heat shock proteins (Hsps) that rescue cells from injury [1-3]. The major heat shock protein 70 (Hsp70) and other Hsps may protect against toxic proteins that develop during neurodegenerative diseases. The Hsps prevent incorrect protein interactions, promotion of protein degradation, sequestration of toxic protein forms, and blockage of signal events that lead to neuronal dysfunction and cell death [2,4]. Interestingly, the induction of Hsps by a stressful stimulus transiently prevents the cell damage by a subsequent and potentially lethal insult of similar or differing origin or intensity. In the nervous system, HS was found to induce the synthesis of Hsp70 mainly in glial cells and less in some neuronal populations [5-8]. In conditions where Hsps capacity is overwhelmed by the excessive amount of unfolded protein molecules, the cells activate a particular sort of stress response called endoplasmic reticulum stress or unfolded protein response (UPR). The accumulation and deposition of misfolded proteins is one of the leading causes of various neurodegenerative diseases [2,4].
Administration of KA to rodents is a potent inducer of excitotoxic damage in the limbic brain structures along with epileptic seizure [9-11]. The neurodegeneration caused by KA is characterized by deterioration of cell perikarya and their processes on the way to cell death [9]. The excitotoxic degeneration is associated with acute expression of many genes, including immediate early gene (IEG) and Hsps genes [12-14]. The morphological and neurochemical damage changes appear clearly distinguishable by 2 h after KA icv injection and become strictly localized to Cornu Ammonis region 3 (CA3) area of the hippocampus [9].
Stress tolerance is a phenomenon where a prior sub-lethal insult leads to protection against a subsequent severe insult. If the mild stress preceded the severe stress of the same type, it might lessen the damage of an organism or cells and the newly attained state of stress resistance is called stress tolerance. Achieving the stress tolerance state of severe stress later is called stress cross-tolerance. Preconditioning neural cells with mild HS induces an HS tolerance, both in vivo and in vitro. Also, mild HS induces cross-tolerance to other types of stress like ischemia, trauma, and epilepsy [3]. There are only a few reports on HS preconditioning of excitotoxic brain damage emphasizing the role of Hsp70. The overexpression of Hsp70 by genetic manipulation provided for protection against many different types of brain damage both in vivo and in vitro [2]. Only exceptionally, however, increasing the Hsp70 levels in neurons of cultured hippocampal slices produced the results that seemed to contradict its role in protection [15]. As compared to Hsp70 induction in HS situation, the Hsp70 induction following the excitotoxic/seizure preconditioning is delayed by 12-24 hours similar to the induction caused by low molecular Hsp70 inducers [2]. Also, there is a major difference in cellular localization of induced Hsp70 following HS, which is mainly in neuroglia, vessels, and some neurons [2,5,7,16]. HS preconditioning produced increased levels of heat-induced Hsp70 in the brain few hours after the whole body hyperthermia (WBH) [7,16,17]. Mild heat exposure for preconditioning the excitotoxic brain damage produced a considerable protection of hippocampal CA3 neurons by preconditioning HS either after two days in the cultured slices or after three days in the hippocampus while there was no apparent protection against KA-induced damage in slices 24 h after KA [18]. Later Duveau group [19] confirmed the presence of Hsp70 in the hippocampal tissue homogenate at 6 h after HS. However, the delayed stage of HS protection seemed to correlate more with the sialylation of Neural Cell-Adhesion Molecule (NCAM) than with Hsp70 induction [19].
Up-to-date, there is no report on the early stage of crosstolerance induced by HS preconditioning on the excitotoxic neuronal damage. However, the very recent report on HS preconditioning of heat-induced damage in nematode Caenorhabditis elegans showed that rapid induction of small Hsps by the mild heating procedure can rescue the C. elegans neurons against threatening heat-induced cell necrosis [20]. If the hypothesis of HS-induced early preconditioning is based on the protective action of Hsp70/Hsps, the time-window to find it is restricted to several hours after the induced stress proteins achieved the steady state in the neural cells of the brain. This study attempted to find if HS-induced crosstolerance to KA-induced neuronal damage also had an early stage and if Hsp70/Hsps should cause it.
Methods
General
Young female Sprague-Dawley (SD) rats weighing 160-200 g (about two months old) were used throughout the experiments. All experiments were conducted in compliance with Guidelines for Animal Experiments of the Animal Resource Facility at the Health Sciences Center of Kuwait University and conformed to National Institutes of Health Guide for Care and Use of Laboratory Animals. They were fed ad libitum with water and standard chow food.
Urethane, a long-lasting anesthesia, was used throughout these experiments. Urethane has little effects on the respiratory system and is the frequently used anesthetic in electrophysiological experiments [21,22]. All animals were sacrificed under the effect of deep anesthesia, and the materials contacted with the anesthetic or rats body fluids were handled with special care. After 4, 24, and 72 hours of hyperthermic treatment, rats were injected with urethane (1.5 g/kg) and placed in Kopf’s stereotaxic apparatus.
We used WBH (41.5°C X 30 min), which represents a mild heat stress, for heat-preconditioning of excitotoxic insult. In the morning hours, slightly anesthetized rats (urethane 0.75 g/kg) were subjected to preconditioning WBH by placing them in a climatic chamber in ventilated cage, with an initial chamber temperature of 43.5°C to reach the target temperature. After 30 minutes of HS planned exposure, the rats were removed from the climatic chamber and were left to cool down till they reach the temperature of 37.5°C. After recovery from anesthesia, they were transferred to the animal room. In all experiments, the control and experimental groups contained six rats. However, in the dose-response experiments only five rats were used per group.
During the icv administration, the rat’s core temperature was monitored with thermostatic control unit set controlling heating pad (Letica Scientific Instruments) with the rectal temperature probe inserted into the anal canal (5-6 cm). The switch-off set point was kept at 37.6°C, which effectively prevented a decrease of body temperature below 37.2°C. Since some rats reached the temperature above the set point due to the effect of drug administration, care was taken to cool the body with the ice-filled bag until it reached the set point temperature. Next, rats were allowed to cool down and acquire normal body temperature before transferring them to the animal room.
KA and drugs were dissolved in sterile saline (PBS: phosphate buffered saline, pH 7.35, 10 mM-Na-K- phosphate buffer). PES/pifithrin was first solubilized in dimethyl sulfoxide (DMSO) and mixed with PBS. All drugs were supplied by Sigma- Aldrich if not stated otherwise. Behavioral signs (whiskering) of KA-induced epileptic activity were registered and evaluated [23]. Accidental movements of the head, fore- and hind-paws were also recorded in the protocol. The anesthetized rats were observed for up to 4 hours after KA induction before sacrifice.
Dose response
This study was based on using a model of KA-induced excitotoxic damage in hippocampal CA3 [9]. The excitotoxic insult was produced in the anesthetized rats that were injected icv with KA. At 4 h post-injection, a selective morphological and biochemical damage appeared in CA3a region of the hippocampus [9]. Based on this, we started testing three different doses of KA (0.6, 0.15 and 0.05 μg) administered icv in three groups of rats to determine the dose of KA required to induce the damage to CA3 area.
Excitotoxicity preconditioning after WBH
After selecting the lowest effective dose of KA, we tested the excitotoxicity preconditioning of the rats at 4 h, 24 h, and 72 h after WBH (41.5°C X 30 min). Then, rats were left to cool down acquiring normal body temperature and either injected with KA after 4 hours interval or transferred to an animal room and injected later at 24 h or 72 h.
Blocking Hsp70 by quercetin and PES/pifithrin μ
The activation of heat shock factor-1 (HSF1) was selectively blocked by quercetin (Q) to demonstrate the role of Hsps in WBH-induced protection as described by Duveau and coworkers [11]. Quercetin (5 mg/kg) was injected I.P. twice, 1 hour before and after exposure to WBH. In other series of experiments, the Hsp70 was blocked by specific Hsp70 inhibitor pifithrin μ. Rats were given an icv injection of 50 μg of pifithrin μ one hour before KA.
Intracerebroventricular injection technique of KA and drugs
KA was injected icv 4 h, 24 h or 72 h after the rat endured HS as described previously [9] and then, after 4 hours, the rat was re-anesthetized with urethane and perfused with fixative. Briefly, after the head of an anesthetized rat was fixed in a Kopf’s stereotaxic frame, the subcutaneous tissue over bregma was anesthetized with local anesthetic (0.1 ml of Xylocaine 2%, Astra, Sweden). After a few minutes, the calvaria was exposed and punctured by a hand-driven penetrator to ensure an icv injection took place with coordinates -0.5 mm posterior to the bregma and 1.3 mm left to the midline. A 30 Gauge needle with ten μl Hamilton syringe fixed in the stereotaxic frame was then slowly inserted into the brain reaching the left lateral ventricle at 4 mm beneath skull surface. KA or drug-containing solution was then injected at a rate of 1.0 μl per minute. Following icv injection, the needle was left in the ventricle for further 5 minutes to prevent reflux [9].
Brain fixation by cardiac perfusion, brain dissection, and parcellation
Animals were deeply anesthetized and were perfused transcardially with saline for two minutes, followed by 100 ml of 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4). The brains were dissected from the skull, parcellated within an adult rat brain mold and kept in the fixative solution overnight at room temperature (RT) for 18-20 hours. On the following day, brain blocks containing the dorsal hippocampus from position -2.5 to -4.5 mm from bregma were sectioned by vibratome (Vibratome 3000, Vibratome, U.S.A.) at 50 microns thickness and transferred to PBS with glycine (200 mg/l). Selected sections were processed for immunohistochemistry (IHC) or Nissl staining with thionin.
Immunohistochemistry
The sections were processed for c-Fos, Hsp70, Hsc70, microglia, and oligodendrocytes IHC [7,9]. Staining with thionin served as visualization of the area of neuronal damage (ND). C-Fos expression (CF) was used as a marker of neuronal activation and biochemical cell damage. IHC stained sections were evaluated quantitatively by image analysis. Alternative sections were used for IHC procedures, with at least six sections from each rat.
Section Analysis
For qualitative analysis, staining with thionin served as a representative morphologic estimation of the extent of the neuronal damage. Thionin staining was used to assess the neuronal morphology with estimation of the degree of degenerative changes [9]. The normal appearance of pyramidal CA3 neurons was demonstrated in rats untreated with KA, whereas a pycnotic appearance of those neurons was displayed in KA-treated animals [9]. To quantitate the extent of neuronal damage (ND) in the pyramidal cell layer of CA3a subregion, the area of the ND (Figure 1) was measured in 6 sections from each animal using a CCD camera equipped microscope with the help of image analysis software (SIS, Germany).
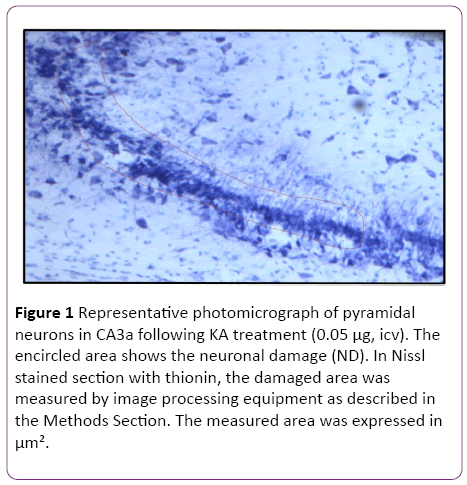
Figure 1: Representative photomicrograph of pyramidal neurons in CA3a following KA treatment (0.05 µg, icv). The encircled area shows the neuronal damage (ND). In Nissl stained section with thionin, the damaged area was measured by image processing equipment as described in the Methods Section. The measured area was expressed in µm².
Quantitative analysis was also performed on the sections stained with the anti c-Fos antibody. C-Fos-positive neurons disappeared in the CA3 regions that were damaged by KAexcitotoxic neurodegeneration. For quantitative analysis, the length of a pyramidal cell layer in CA3a area, which was either showing complete lack or apparent attenuation of c-Fos protein expression, was measured with the same equipment (Figure 2). Detection and counting pyramidal neurons in CA3a area were done following Hsp70 IHC procedure based on ABC technique using Vectastain Elite kit [7], with image analysis of Hsp70 stained neurons in CA3a, b and c sectors [9]. FluoroJade staining of degenerating neurons was performed on sections of control and preconditioned rats as described by Schmued et al. [24].
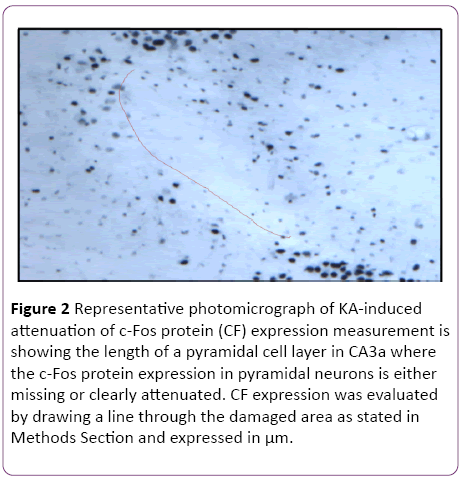
Figure 2: Representative photomicrograph of KA-induced attenuation of c-Fos protein (CF) expression measurement is showing the length of a pyramidal cell layer in CA3a where the c-Fos protein expression in pyramidal neurons is either missing or clearly attenuated. CF expression was evaluated by drawing a line through the damaged area as stated in Methods Section and expressed in µm.
Statistical Analysis
Statistical analysis of quantitative immunohistochemical data was conducted by parametric ANOVA and/or nonparametric Kruskal-Wallis test using SPSS® statistical program (v19, SPSS Inc., Chicago, IL, USA). Statistically significant differences between individual treatments were evaluated with post-hoc Bonferroni's analysis of data or with Man- Whitney test as appropriate. The differences were considered statistically significant if they reached a P value ≤0.05.
Results
Dose response of the kainate that could be effectively produced protection from heat stress
In the initial part of this study, experiments were designed to find the appropriate dose of KA, which if injected into the standard heat stress-pretreated rat could produce the reduction in neural damage and attenuation of c-Fos protein expression in the CA3a area of the hippocampus. It is known that a KA dose of 0.15 μg icv induces a typical ND in the hippocampal CA3a, but rarely affects the adjacent areas CA3b or CA3c [9]. The higher (0.6 μg) and lower (0.05 μg) doses of KA were, therefore, selected accordingly. As for the time interval between HS and KA icv, an interval of 72 h was chosen for these dose-response experiments based on the report that significant protection of CA3 neurons by HS began only after three days following HS [18]. The neural damage found after injecting any doses (0.6, 0.15 and 0.05 μg) of KA was very apparent. Figure 1 shows an example of pyramidal neurons in CA3a following 0.05 μg KA treatment. In fact, the dose of 0.6 μg did extensive damage to the whole area CA3, sometimes reaching the hilus of the dentate gyrus (data not shown). Likewise, Figure 2 displays the length of a pyramidal cell layer in CA3a where the c-Fos protein expression in pyramidal neurons is either missing or clearly attenuated following KA icv injection. However, our observations showed that the neuronal damage induced by kainate was remarkably decreased by hyperthermia with 0.05 μg KA dose (Figure 3A) compared to 0.15 μg (Figure 3B) and 0.6 μg (Figure 3C) KA doses. Analysis of the kainate-induced ND in the hippocampus following the different dosages of kainite injections (0.05, 0.15 and 0.6 μg) showed that 0.05 μg KA injection significantly (P<0.05) attenuated the ND in the CA3a area (Figure 3D) after hyperthermic treatment. Likewise, the immunohistochemical staining of the c-Fos protein expression in the CA3a pyramidal neuronal layer following 0.05 (Figure 4A) and 0.15 (Figure 4B) μg of KA dosage revealed a noticeable attenuation of c-Fos protein expression in the 0.05 μg of KA-injected rats following hyperthermic treatment. Analysis of the hyperthermic protection of KA-induced attenuation of c-Fos protein expression showed significantly (P<0.05) decrease in 0.05 μg KA-injected rats compared to 0.15 μg KA-injected group (Figure 4C). Thus, the damage induced by kainate was significantly decreased by hyperthermia when the KA dose was 0.05 μg (Figures 3 and 4), and this applies to both evaluated parameters ND (77%) and CF (75%). Therefore, the dose of 0.05 μg KA was used in the rest of the planned experiments.
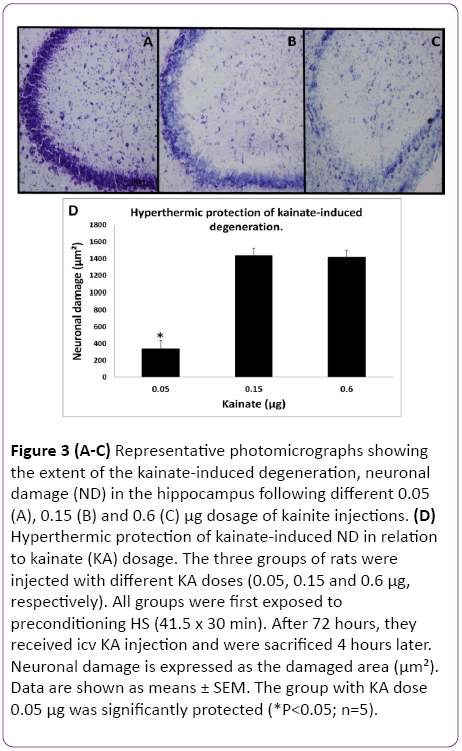
Figure 3: (A-C) Representative photomicrographs showing the extent of the kainate-induced degeneration, neuronal damage (ND) in the hippocampus following different 0.05 (A), 0.15 (B) and 0.6 (C) µg dosage of kainite injections. (D) Hyperthermic protection of kainate-induced ND in relation to kainate (KA) dosage. The three groups of rats were injected with different KA doses (0.05, 0.15 and 0.6 µg, respectively). All groups were first exposed to preconditioning HS (41.5 x 30 min). After 72 hours, they received icv KA injection and were sacrificed 4 hours later. Neuronal damage is expressed as the damaged area (µm²). Data are shown as means ± SEM. The group with KA dose 0.05 µg was significantly protected (*P<0.05; n=5).
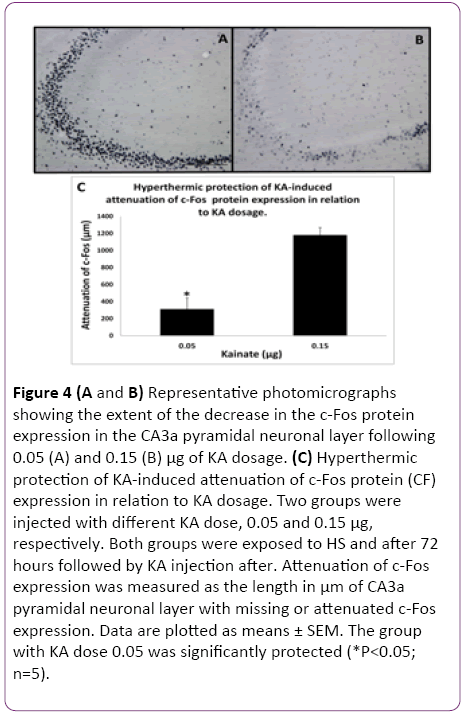
Figure 4: (A and B) Representative photomicrographs showing the extent of the decrease in the c-Fos protein expression in the CA3a pyramidal neuronal layer following 0.05 (A) and 0.15 (B) µg of KA dosage. (C) Hyperthermic protection of KA-induced attenuation of c-Fos protein (CF) expression in relation to KA dosage. Two groups were injected with different KA dose, 0.05 and 0.15 µg, respectively. Both groups were exposed to HS and after 72 hours followed by KA injection after. Attenuation of c-Fos expression was measured as the length in µm of CA3a pyramidal neuronal layer with missing or attenuated c-Fos expression. Data are plotted as means ± SEM. The group with KA dose 0.05 was significantly protected (*P<0.05; n=5).
Effect of HS preconditioning on KA-induced changes as a function of interval between HS and KA injection
Since the reported hyperthermic induction of Hsp70 protein in the rat hippocampus achieved significant presence between 1.5 and 4 hours, we decided to include a 4 h interval after HS as the first time interval to investigate its effect on protection of KA-induced damage. The results of early (4 h) and late preconditioning (24 h and 72 h) intervals between HS-treated and KA-injected groups and the control rats, which were not subjected to HS but treated with KA alone are shown in Figures 5 and 6. A remarkable decrease in the neurodegeneration in the hippocampal CA3a was noticed in the HS4, HS24 and HS72 preconditioning before KA injections (Figure 5A-5C) compared to control rats (Figure 5D) injected with KA only. Histological and morphometric analysis showed that the HS4 and HS72 groups had significantly lower ND values as compared to control group (P<0.05), while HS24 rats did not reach the set significance level. Likewise, sections of the c-Fos protein expression in the CA3a pyramidal neuronal layer at early preconditioning (4 hrs; Figure 6A), and late preconditioning (24 h; Figure 6B and 72 h; Figure 6C) before KA injections showed a noticeable decrease compared to KAinjected control animals (Figure 6D). Analysis of the c-Fos protein staining in the CA3a hippocampal area (Figure 6E) revealed significant (P<0.05) decrease in the HS4 and HS72 groups while HS24 rats did not reach the set significance level.
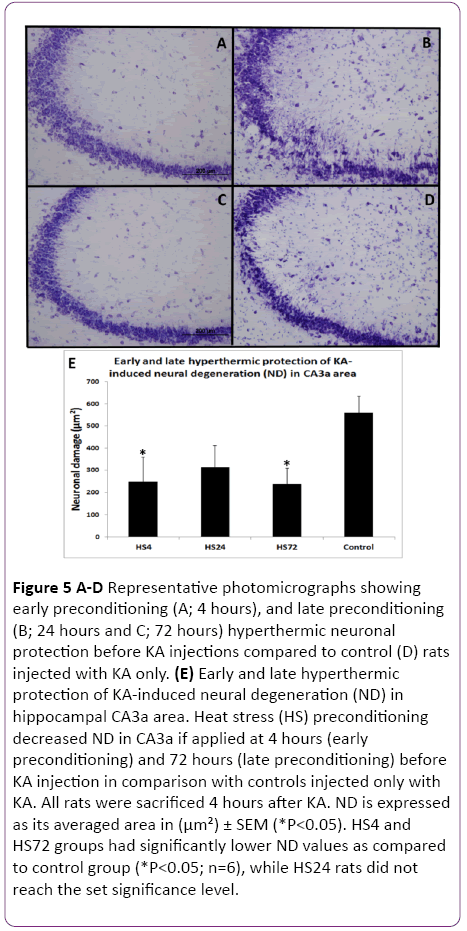
Figure 5: A-D Representative photomicrographs showing early preconditioning (A; 4 hours), and late preconditioning (B; 24 hours and C; 72 hours) hyperthermic neuronal protection before KA injections compared to control (D) rats injected with KA only. (E) Early and late hyperthermic protection of KA-induced neural degeneration (ND) in hippocampal CA3a area. Heat stress (HS) preconditioning decreased ND in CA3a if applied at 4 hours (early preconditioning) and 72 hours (late preconditioning) before KA injection in comparison with controls injected only with KA. All rats were sacrificed 4 hours after KA. ND is expressed as its averaged area in (µm²) ± SEM (*P<0.05). HS4 and HS72 groups had significantly lower ND values as compared to control group (*P<0.05; n=6), while HS24 rats did not reach the set significance level.
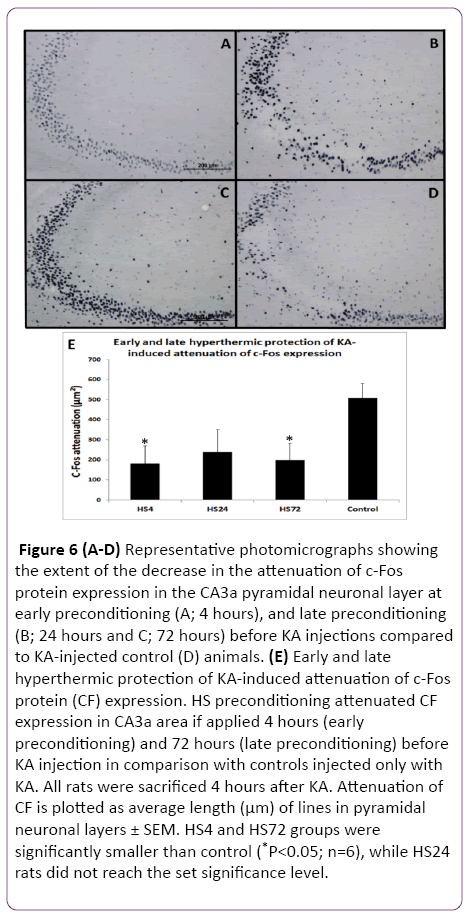
Figure 6: (A-D) Representative photomicrographs showing the extent of the decrease in the attenuation of c-Fos protein expression in the CA3a pyramidal neuronal layer at early preconditioning (A; 4 hours), and late preconditioning (B; 24 hours and C; 72 hours) before KA injections compared to KA-injected control (D) animals. (E) Early and late hyperthermic protection of KA-induced attenuation of c-Fos protein (CF) expression. HS preconditioning attenuated CF expression in CA3a area if applied 4 hours (early preconditioning) and 72 hours (late preconditioning) before KA injection in comparison with controls injected only with KA. All rats were sacrificed 4 hours after KA. Attenuation of CF is plotted as average length (µm) of lines in pyramidal neuronal layers ± SEM. HS4 and HS72 groups were significantly smaller than control (*P<0.05; n=6), while HS24 rats did not reach the set significance level.
The results of these experiments indicated that there was a significant difference between the HS preconditioned, and control rats at 4 h and 72 h intervals (Figures 5 and 6). ND and CF parameters decreased by 62% and 64% in 4 hrs animals (P<0.05), respectively, and by 64% and 60% in 72 h groups (P<0.05), respectively. However, in the 24 h interval animals, the evaluation of ND and CF did not show a significant difference despite the clear tendency for decreasing their values in comparison to controls (Figure 5E and Figure 6E).
Elucidation the role of Hsps in heat-induced cross-tolerance by quercetin
The protective effect of HS on heat-induced tolerance and cross tolerance is mainly ascribed to induced Hsp70 or other Hsps. To elucidate the role of Hsp70/Hsps in WBH-induced protection, the activation of HSF1 was blocked by quercetin (Q) as a useful inhibitor of Hsps induction. HSF1, a heatinducible transcription factor present in the cytosol, is released by acute HS from its complex with Hsp90 and is translocated to the nucleus where it initiates transcription of Hsp70/Hsps genes. Animals treated with Q blocking at 4 h (Figure 7B) showed remarkable ND attenuation in the CA3a pyramidal neuronal layer compared to 4 h Q-untreated group (Figure 7A) following hyperthermic protection of KA induction. Meanwhile, there was no noticeable difference between Qtreated (Figure 7D) and Q-untreated (Figure 7C) at 72 h after hyperthermic protection of KA. Analysis of tissue sections for ND (Figure 7E) showed that administration of Q before and after the preconditioning heat stress resulted in a significant (P<0.05) increased KA-induced neural damage after 4 h in comparison with the animals, which were preconditioned but did not receive Q injections. There was no significant difference between Q-treated and Q-untreated rats in the KAinjected at 72 h after HS. Similarly, the c-Fos-immunoreactive staining in the CA3a pyramidal neuronal layer was remarkably abundant due to quercetin blocking at the 4 h (Figure 8B) interval following hyperthermic protection of KA induction compared to control 4 h (Figure 8A) animals.
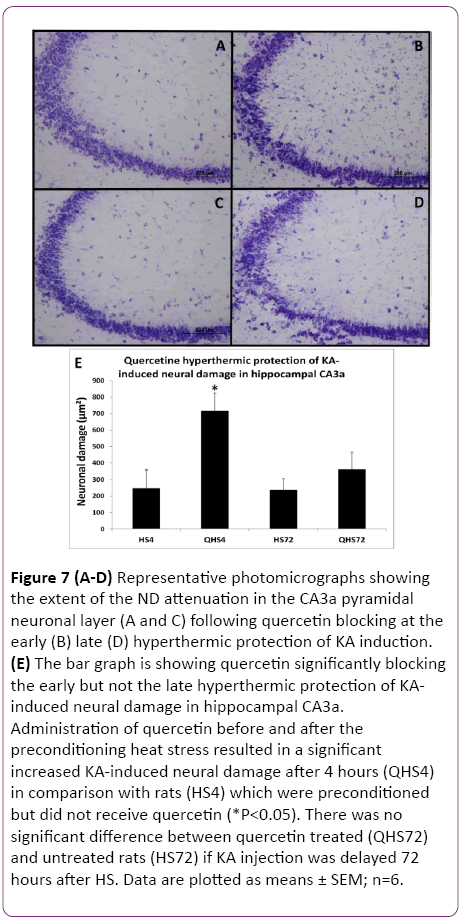
Figure 7: (A-D) Representative photomicrographs showing the extent of the ND attenuation in the CA3a pyramidal neuronal layer (A and C) following quercetin blocking at the early (B) late (D) hyperthermic protection of KA induction. (E) The bar graph is showing quercetin significantly blocking the early but not the late hyperthermic protection of KAinduced neural damage in hippocampal CA3a. Administration of quercetin before and after the preconditioning heat stress resulted in a significant increased KA-induced neural damage after 4 hours (QHS4) in comparison with rats (HS4) which were preconditioned but did not receive quercetin (*P<0.05). There was no significant difference between quercetin treated (QHS72) and untreated rats (HS72) if KA injection was delayed 72 hours after HS. Data are plotted as means ± SEM; n=6.
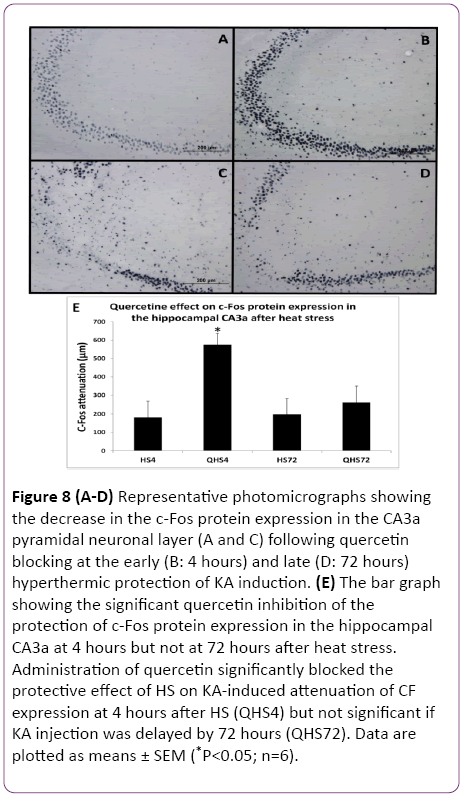
Figure 8: (A-D) Representative photomicrographs showing the decrease in the c-Fos protein expression in the CA3a pyramidal neuronal layer (A and C) following quercetin blocking at the early (B: 4 hours) and late (D: 72 hours) hyperthermic protection of KA induction. (E) The bar graph showing the significant quercetin inhibition of the protection of c-Fos protein expression in the hippocampal CA3a at 4 hours but not at 72 hours after heat stress. Administration of quercetin significantly blocked the protective effect of HS on KA-induced attenuation of CF expression at 4 hours after HS (QHS4) but not significant if KA injection was delayed by 72 hours (QHS72). Data are plotted as means ± SEM (*P<0.05; n=6).
The c-Fos immunoreactivity was much less apparent in both. In contrast, there was no observable difference between Quntreated (Figure 8C) and Q-treated (Figure 8D) at 72 h after hyperthermic protection of KA. Morphometric analysis (Figure 8E) revealed that administration of Q significantly (P<0.05) blocked the protective effect of HS on KA-induced attenuation of CF expression at 4 h after HS but not when 72 h delayed KA injection. The evaluation of ND and CF expression showed that Q abolished the effect of HS preconditioning only in rats with a 4 h interval between HS and KA. Both measurements of ND (Figure 7) and CF (Figure 8) in these rats reached significantly (P<0.05) increased values as compared to the controls by 65% and 66%, respectively. However, the preconditioning effect was still present after a 72 h interval and apparently was not influenced by Q blocking. Therefore, blocking Hsp70/Hsps expression with Q abolished the protection at 4 h but not at 72 h.
Blocking Hsp70 action by pifithrin μ (2- Phenylethynesulfonamide -PES)
Since the Q blocking experiments indicated that either Hsp70 alone or some other Hsps may be involved in the observed HS-induced protection of hippocampal ND and CF, the selective inhibitor of Hsp70 action PES was used. Following HS preconditioning, PES was injected icv three hours after HS (one hour before KA) so that the 4 h interval between HS and KA was maintained. The control rats were treated as previously described in the methodology. The histological staining assessment of the effect of the PES on the ND at early phase (4 h) following the KA injection and HS treatment in CA3a area of the hippocampus (Figure 9B) did not show any remarkable change compared to HS-treated+KA-injected (Figure 9A) and PES-treated+KA-injected (Figure 9C) control groups. Statistical analysis of HS+KA and HS+PES+KA groups showed that PES did not block the HS-induced early protection of KA-induced neuronal damage (Figure 9D). PES+KA rats were only treated with a same dose of PES and followed by KA injection 60 min later. Surprisingly, PES also blocked the neuronal damage caused by KA injection. Likewise, the PES injection did not show any noticeable change in the c-Fos immunostaining at early phase (4 h) following KA injection in CA3a area of the hippocampus (Figure 10B) compared to heat shock treated (Figure 10A) and PES-treated (Figure 10C) control KA-injected groups. Analytical comparison of HS+KA and HS+PES+KA groups (Figure 10D) showed that PES did not block the HSinduced early protection of KA-induced attenuation of CF expression. PES+KA rats were only treated with PES and followed by KA injection 60 min later. PES also blocked the attenuation of CF expression caused by KA injection.
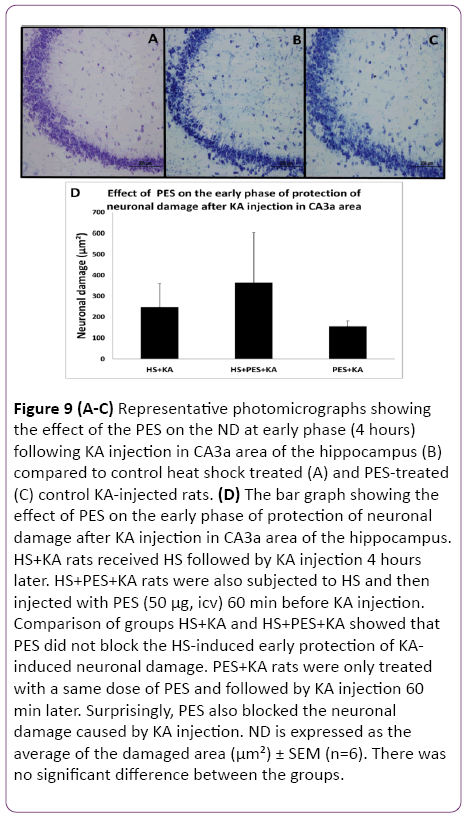
Figure 9: (A-C) Representative photomicrographs showing the effect of the PES on the ND at early phase (4 hours) following KA injection in CA3a area of the hippocampus (B) compared to control heat shock treated (A) and PES-treated (C) control KA-injected rats. (D) The bar graph showing the effect of PES on the early phase of protection of neuronal damage after KA injection in CA3a area of the hippocampus. HS+KA rats received HS followed by KA injection 4 hours later. HS+PES+KA rats were also subjected to HS and then injected with PES (50 µg, icv) 60 min before KA injection. Comparison of groups HS+KA and HS+PES+KA showed that PES did not block the HS-induced early protection of KAinduced neuronal damage. PES+KA rats were only treated with a same dose of PES and followed by KA injection 60 min later. Surprisingly, PES also blocked the neuronal damage caused by KA injection. ND is expressed as the average of the damaged area (µm²) ± SEM (n=6). There was no significant difference between the groups.
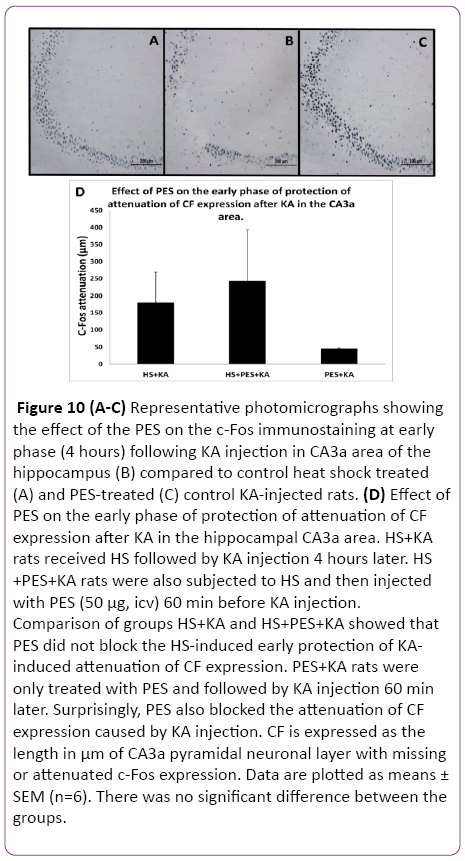
Figure 10: (A-C) Representative photomicrographs showing the effect of the PES on the c-Fos immunostaining at early phase (4 hours) following KA injection in CA3a area of the hippocampus (B) compared to control heat shock treated (A) and PES-treated (C) control KA-injected rats. (D) Effect of PES on the early phase of protection of attenuation of CF expression after KA in the hippocampal CA3a area. HS+KA rats received HS followed by KA injection 4 hours later. HS +PES+KA rats were also subjected to HS and then injected with PES (50 µg, icv) 60 min before KA injection. Comparison of groups HS+KA and HS+PES+KA showed that PES did not block the HS-induced early protection of KAinduced attenuation of CF expression. PES+KA rats were only treated with PES and followed by KA injection 60 min later. Surprisingly, PES also blocked the attenuation of CF expression caused by KA injection. CF is expressed as the length in µm of CA3a pyramidal neuronal layer with missing or attenuated c-Fos expression. Data are plotted as means ± SEM (n=6). There was no significant difference between the groups.
Immunohistochemistry of Hsp70 in hippocampal sections following HS
The results of CA3a area immunohistochemical staining of the HS-induced Hsp70 protein expression at 4 h, 24 h and 72 h intervals following HS revealed that the distribution of Hsp70 immunoreactivity from animals with 4 h interval following the HS occurred in small cells and vessels (Figure 11A). Apparently, there were no large pyramidal neurons stained by the anti- Hsp70 antibody. The higher magnification micrographs (Figure 11B) confirmed that the expressed Hsp70 was mainly localized in the nuclei of small cells, presumably neuroglia, and in walls and endothelial cells of the vasculature (Figure 11C). While the absence of neuronal Hsp70 immunostaining by hyperthermia in the hippocampus was typical for all selected time points, there were neural structures in the vicinity of the hippocampal formation that showed a considerable quantity of Hsp70 immunoreactivity in their neuronal populations. The neurons of the medial habenula nucleus were profoundly immunostained with anti-Hsp70 protein antibody at 4 h after hyperthermic treatment (Figure 12A and 12B). However, its content declined at 24 h post-HS (Figure 12C) and completely disappeared by 72 h after HS as well as from the glial and vascular cells of CA3a area (Figure 12D). Since this nucleus is quite close to the hippocampal formation, Hsp70 expression in the neurons of the medial nucleus of habenula was considered an indicator of efficient HS procedure in inducing the Hsp70. However, the expression of Hsp70 protein in longer intervals examined following HS rapidly decreased after the 24 h interval, and at a 72 h interval. The controls were incubated without primary anti-Hsp70 antibody and did not show any significant staining (not shown).
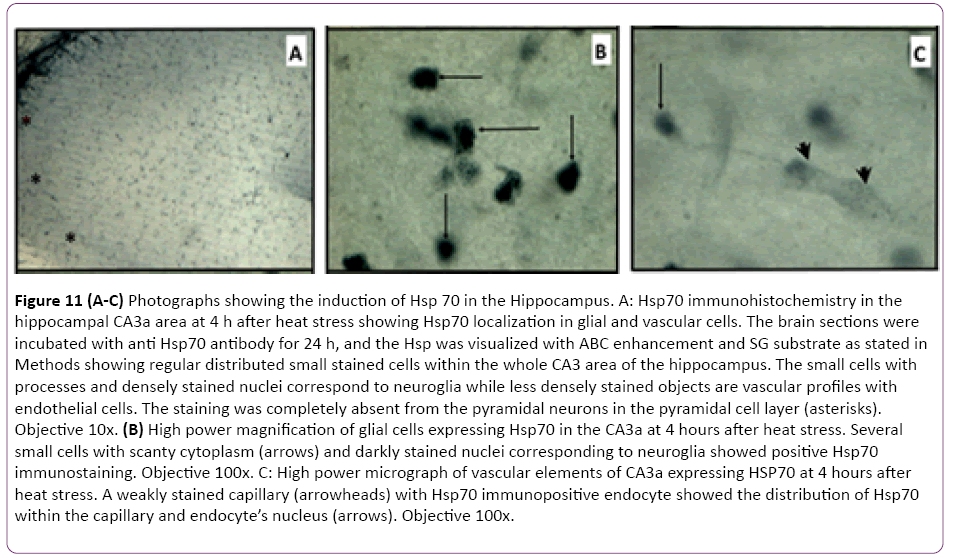
Figure 11: (A-C) Photographs showing the induction of Hsp 70 in the Hippocampus. A: Hsp70 immunohistochemistry in the hippocampal CA3a area at 4 h after heat stress showing Hsp70 localization in glial and vascular cells. The brain sections were incubated with anti Hsp70 antibody for 24 h, and the Hsp was visualized with ABC enhancement and SG substrate as stated in Methods showing regular distributed small stained cells within the whole CA3 area of the hippocampus. The small cells with processes and densely stained nuclei correspond to neuroglia while less densely stained objects are vascular profiles with endothelial cells. The staining was completely absent from the pyramidal neurons in the pyramidal cell layer (asterisks). Objective 10x. (B) High power magnification of glial cells expressing Hsp70 in the CA3a at 4 hours after heat stress. Several small cells with scanty cytoplasm (arrows) and darkly stained nuclei corresponding to neuroglia showed positive Hsp70 immunostaining. Objective 100x. C: High power micrograph of vascular elements of CA3a expressing HSP70 at 4 hours after heat stress. A weakly stained capillary (arrowheads) with Hsp70 immunopositive endocyte showed the distribution of Hsp70 within the capillary and endocyte’s nucleus (arrows). Objective 100x.
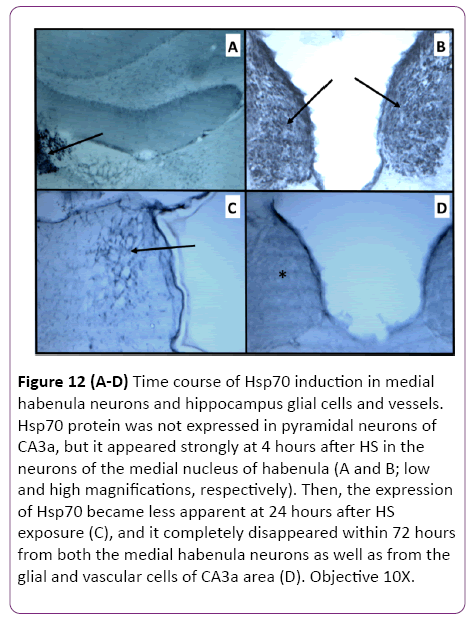
Figure 12: (A-D) Time course of Hsp70 induction in medial habenula neurons and hippocampus glial cells and vessels. Hsp70 protein was not expressed in pyramidal neurons of CA3a, but it appeared strongly at 4 hours after HS in the neurons of the medial nucleus of habenula (A and B; low and high magnifications, respectively). Then, the expression of Hsp70 became less apparent at 24 hours after HS exposure (C), and it completely disappeared within 72 hours from both the medial habenula neurons as well as from the glial and vascular cells of CA3a area (D). Objective 10X.
Discussion
Hsp70 and IEG c-Fos and c-Jun are the most frequently used cellular stress indicators in various conditions [25]. IEG induction can regulate the expression of late target response genes that could produce long-term changes in cells [26-29]. CFos has a presumed role, in general, stress reaction to physical or psychological stressors [29]. C-Fos expression in acute stress has been used as a functional marker to visualize synaptically and/or transcriptionally activated neurons in various regions of the brain [26,30,31].
Heat stress and confirmation of Hsp70 induction
HS is a well-established procedure to attain a state of increased resistance to more profound, even damaging HS insult, which is labeled either as HS tolerance (mild HS preconditioning of severe HS consequences) or cross-tolerance (mild HS preconditioning of consequences of another severe stress type). In this study, we investigated the possibility to protect neuronal and biochemical damages induced by excitotoxic KA in the CA3 region of the hippocampus by preceding exposure of experimental rats to mild heat stress, i.e., HS-induced cross tolerance of excitotoxic damage. While general anesthesia of rats might be a complicating factor, it provided a possibility to standardize the heating procedure in the climatic chamber and eliminated additional stress influences that are unavoidable in non-anesthetized rats [32]. The used protocol (HS for 30 min at 41.5°C) induced clearly detectable Hsp70 protein in the hippocampus with the typical cellular distribution. We used specifically Hsp70 protein expression in the neurons of the medial nucleus of habenula, which if present, provided unequivocal proof of Hsp70 expression since these neurons belong to the class of forebrain neurons with most notable heat-induced Hsp70 expression [6,7]. Since there are inherent limitations in exploiting stronger hyperthermia (>42°C) for the purpose of increasing Hsp70 production in brain cells of experimental animals, the rapidly falling levels of mild heat-induced Hsp70 attenuated gradually the possibility to detect the existence of heat-induced crosstolerance in later time intervals [18].
There are two distinct stages of HS preconditioning (or induced cross-tolerance), the early stage, and the late stage. Four hours after HS preconditioning detected the early stage of HS-induced cross-tolerance to the damaging effect of KA. Then, there was a transition to a delayed stage of crosstolerance that became apparent by 72 h after HS. However, there was not a clear difference between these two stages which would be observable in Nissl stained and c-Fos immunostained brain sections. On the contrary, it was the noticeable absence of Hsp70 protein immunostaining that contrasted with the presence of Hsp70 immunostaining in the 4 h-interval rat brain (Figure 12C and 12D). The induction of the Hsp70 at 4 h and its disappearance at 72 h in medial habenula neurons as well as in glial and vascular cells means that there is another factor(s) that may cause a protection in the late stage (72 h) other than the Hsp70.
Model of the selective neural damage induced by excitotoxic insult
The icv injection of KA into one ventricle, as compared to systemic injection, was more efficient, since only submicrogram dosage was necessary to produce a typical neural damage and biochemical disturbance [14,18,22]. Selecting the lowest effective dose of KA (0.05 μg) elicited typically, circumscribed lesion of CA3a area of the ipsilateral hippocampus. It provided for morphological damage of pyramidal neurons and their accompanying attenuation of c- Fos protein expression within the borders of CA3a area. The extreme sensitivity of CA3a area of the hippocampus is due to the accumulation of high-affinity AMPA/KA postsynaptic receptors on dendrites and bodies of pyramidal cells in CA3a, and presynaptic receptors on the mossy fibers [22,33]. The extension of the area of neuronal damage to adjacent areas CA3b and CA3c could be seen in studies using higher doses of icv KA and may be a source of additional variability in the results [18,34,35]. Since the ND of pyramidal neurons developed very fast after KA injection, the 4 h interval between the KA injection and the sacrifice of the rat was considered suitable both for evaluation of ND as well as for attenuation of CF expression [9]. Also, this interval provided for prevention of unnecessary suffering and pain of animals recovering later from anesthesia.
The morphological changes of the Nissel-stained pyramidal neurons in ND area (aberrant neuronal shape, abnormal staining density, pycnotic appearance, loss of dendrites, and even the whole neurons) were clearly distinguishable. They corresponded to apoptotic and necrotic processes, which were confirmed by previous electron microscopic studies [9,10]. It is not possible to exclude that some of the affected pyramidal neurons in CA3a area were capable of partial/complete recovery later since the determination of ND area encompassing damaged pyramidal neurons could not be equalized with an area of total cell destruction. Similar to ND changes, the profound changes in ability to express c-Fos protein following KA-injection were easily evaluated since most damaged pyramidal neurons completely lost the visible c-Fos immunostaining.
KA dose-response in relation to HS-induced protection
For the estimation of the effective dose of KA- inducing neural damage, which could be protected by used HS, we tested three doses of KA (0.6, 0.15 and 0.05 μg). Then, we used the 72 h interval between the HS and KA injection, since it was reported that HS-induced cross-tolerance could be induced by delaying the interval between HS and KA injection by at least 72 hours, both in vivo and in-vitro [18]. The effective induction of ND and attenuation of CF expression was first established in controlled anesthetized rats that received only 0.05 μg of KA icv and were sacrificed 4 h later. Exposing rats to HS and injecting them with this low dose of KA resulted in significant protection of both measured parameters. However, the HS failed to induce the expected protection of KA-induced damage at the higher KA dosages (0.15 and 0.6 μg). Our results are in contrast to the Duveau et al. [18] study that showed 0.6 μg KA significantly protected the neural damage when measured seven days after KA insult. Despite the apparent similarity in the experiments of our study and Duveau et al., [18] study, there are some substantial differences. Duveau and coworkers used free moving nonanesthetized rats that might have reacted to the heat in a different way than our anesthetized rats. Then, the evaluation of surviving numbers of neurons in the whole CA3 region might be less sensitive to changes in the most responsive subregion of CA3, which is CA3a. Our study found that CA3a is most affected by KA, since it has the highest density of highaffinity KA receptors [22,33]. Counting the surviving neurons in the whole CA3 may evaluate the protective effect of HS on CA3b or CA3c neurons while CA3a neurons might be completely extinguished, despite the observed protection in other sub-regions [18]. Delaying the evaluation of neural damage by 24 hours or more has to take into consideration that some damaged pyramidal neurons express Hsp70 [12,16,36]. Finally, entirely different mechanism of protection may work in these two experimental designs [19].
Early and late HS-induced cross-tolerance to excitotoxic insult
To the best of our knowledge, our results show for the first time the existence of early stage of HS-induced tolerance to excitotoxic insult. The 4 h interval provides for the coincidence of real expression of Hsp70 in hippocampus and tolerance to KA-induced ND and attenuation of CF protein expression, which was the major hypothesis of our study. Early induction of Hsp70 by heat in CA3a was observed by many other studies [5-7,16]. The appearance of Hsp70 protein in the brain is, however, slower in comparison with cells in vitro or some other organs, since the brain is protected by brain cooling system [37] and the intensity of heating the brain is limited because of heatstroke [38].
The experiments using 24 h interval did not show a significant difference between HS-treated and control rats, but there was a tendency to achieve the protected ND and CF values. A study on timing of tolerance induction recognized the transition between the early and late/delayed stages [39]. It showed that the early stage begins after 24 h following the heat exposure because the acute stage of protection was not observed [39]. According to our hypothesis, the decrease of Hsp70 cellular content in CA3a might provide for the transitional stage, when some additional or entirely different mechanism is recruited. The observation of decreasing immunostaining of Hsp70 in the forebrain corresponded to the estimated turnover of Hsp70 protein at 12-24 hours [39].
The late tolerance phase was present in the 72 h interval after HS. However, at this interval the expression of Hsp70 protein could not be detected by the immunohistochemical method in the CA3a. In the indicator structure of medial habenula, it was clearly at the background level. It could be speculated that another mechanism responsible for tolerance to KA-induced damage was activated. This idea is supported by a recent study indicating the recruitment of NCAM sialylationdependent tolerance mechanism [19].
Blocking activation of Hsp70/Hsps by HSF1 inhibitor quercetin
Quercetin data corroborated the view that early phase of observed cross-tolerance may be related to the presence of Hsp70 in the protected structure. This inhibitor of HSF1 activation was used before and after the HS stage, and it blocked the HS-induced protection of KA damage but only when the interval between HS and KA injection was 4 hours. The failure to alleviate the late phase of HS-induced tolerance supported the above speculation that the late phase of HSinduced cross-tolerance was not dependent on the Hsp70 presence in the damaged structure. The results of the Q implied that other factors may be responsible for the delayed phase of heat induced cross tolerance while it strengthens the possibility that Hsp70/Hsps were involved in the induction of cross-tolerance at the early stage.
Hsp70 expression is regulated together with most other Hsps by activation of specific transcription factor HSF1 [40]. The major player in the complex control of HSF1 activation/ deactivation is Hsp90, which with the help of some other chaperones/cochaperones (including even Hsp/Hsc70), keeps HSF1 in an inactive state. Interference at any step of Hsp1 activation, like its release from the complex, phosphorylation or trimerization, is followed by decreased/blocked production of Hsps including Hsp70. In our study, we used a flavonoid Q, which is the most potent inhibitor and less toxic than other flavonoids like luteolin, apigenin or genistein. Despite its frequent use for the inhibition of Hsps including Hsp70 via its blocking of HSF1 phosphorylation, it was also shown to inhibit protein kinases including tyrosine kinases and consequently it could influence indirectly the behavior of many proteins and signaling pathways [41]. It is not, therefore, surprising to find the Q to influence the vast spectrum of cellular effects, e.g., glial deactivation [42] and Hsp70 nuclear translocation [43]. It was also used for blocking HS preconditioning of KA-induced neural damage in the hippocampus, but the study of Duveau and associates concluded that it was rather the blocking of the polysialylation of NCAM than that of Hsp70 which prevented the notable appearance of the late stage tolerance [19].
Direct inhibition of Hsp70 by PES did not attenuate HS-induced phase of early tolerance of KA-induced neural damage
Surprisingly, our results showed that PES did not block the HS-induced protection of early stage of tolerance KA-induced insult. PES was originally identified by its inhibitory action on the direct mitochondrial pathway of p53-mediated apoptosis [44]. Since p53 acts as a transcription factor for proapoptotic proteins Bax, PUMA, and Noxa, its inhibition would prevent the above proteins to be produced [44]. Also, a recent analysis showed that p53 permeabilized the mitochondrial membrane directly and released mitochondrial proapoptotic proteins cytochrome c, and mitochondrial protein referred as a second mitochondrial-derived activator (Smac/DIABLO) into the cytosol [45]. The PES did not prevent the rescue of hippocampal ND as well as CF-induced expression by HS preconditioning. The lack of effect of PES administration on HS-induced early protection could be the consequence of the direct action of PES on the KA-induced neural damage. This possibility was confirmed by injecting the rats with PES icv before KA. This experiment clearly showed that PES prevented the KA-induced neuronal damage in CA3a as well as the attenuation of c-Fos protein. The unexpected results of the failure of PES to suppress HS-induced protection raised a possibility that PES might influence the sole damaging process induced by KA.
Mechanism of the protective effect of HSinduced Hsp70/Hsps in the early stage of tolerance to KA-induced neural damage
The original work from Tytell’s laboratory showed that Hsp70 induced by heat in ensheathing glial cells of squid giant axon was rapidly translocated to adjacent axonal process [46]. The possibility of protective or tolerance inducing effects of Hsp70, which was transported from glia to neurons were summarized by Tytell [47]. Any influence on the transfer of the neural signals between neurons that is considered the basis of neural function could be of utmost importance. The perisynaptic glial processes that cover the pre- and postsynaptic structures could transport and/or release the Hsp70/Hsps produced in glial cells at these sites that are critical for neuron communication [1,48,49]. Such reasoning is adequate in cases when the endogenous capacity of neurons to induce Hsp70/Hsps under hyperthermic treatment is limited [5-7,16]. The hyperthermic time limitation was again confirmed in our study since the induction of Hsp70 in the pyramidal neurons of CA3 subregion of the hippocampus was not detectable by employed IHC technique. On the contrary, there was a significant induction of Hsp70 in the glial cells but only for a limited period that coincided with the early stage of tolerance to excitotoxic insult. We could not exclude the participation of other Hsps, such as small crystalline-like Hsps. The protective effect of sHsp-16.1 induced by heat preconditioning by mildly elevated temperature on the survival and neurodegeneration induced by heat stroke in Caenorhabditis elegans was recently reported [20]. The nematode model of necrotic cell death induced by heat stroke showed that preconditioning at 34°C for 30 min was enough to protect the nematodes against the heat stroke (at 39°C for 15 min) given only 20 min later, confirming thus convincingly the existence of the early stage of tolerance. A similar results were obtained in an in vitro experiments using mammalian embryonic stem cell-derived neurons [20].
Several possibilities may be considered to implicate the Hsp70 in the observed neuronal protection. First, Hsp70 is present in glia from where it might be transferred to neurons [2,47,50,51]. Second, since Hsp70 molecules are present also in vessels, a transfer from vessels (and glia) to neurons is another possibility. Third, extracellular Hsp70 can influence synaptic transmission via an effect on the mossy fibers synaptic transmission to pyramidal neurons [52,53]. Also, other possibilities are not involving Hsp70. The first one is that other Hsps might be involved like small Hsps. The second is that Hsps mechanism might play a role. For example, induction of Golgi ATPase sequestering calcium induced by WBH is preconditioning neurons to calcium influx caused by KAinduced excitotoxicity [20]. Another one is that activation of signaling pathway PI3K-Akt by WBH results directly in the blockade of cell death.
In conclusion, our study has revealed that hyperthermic preconditioning of KA-induced damage protected CA3a neurons when given 4 h (early stress tolerance) or 72 h (late stress tolerance) before the insult. Blockade of Hsp70 expression abolished hyperthermic protection at 4 h but not at 72 h. The same results were obtained for the attenuation of c- Fos expression in CA3a. We suggest that the mechanism of early hyperthermic protection of KA-induced excitotoxic damage involves Hsp70 or other types of Hsps, which are induced in neurons or transferred from glial cells to neurons. On the contrary, the late protection may not require the actual presence of Hsp70/Hsps in CA3a neurons or glial cells. Achieving tolerant state against noxious stress factors by innocuous use of physical or pharmacological means would be an important instrument in disease fighting. It seems that Hsp70 pleomorphic ways of protection against various types of cell death makes this stress protein a desirable means of prevention in ischemic stroke, trauma, hemorrhage, epilepsy, and neurodegeneration.
Acknowledgements
The authors acknowledge the contribution of Mrs. Glory Alexander from Department of Physiology and Mr. Jijin Kumar from Department of Anatomy for their excellent technical assistance in the experiments conducted in this study. We would also like to thank the Graduate School at Kuwait University for their support.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
8680
References
- Bechtold DA, Brown IR (2000) Heat shock proteins hsp27 and hsp32localize to synaptic sites in the rat cerebellum following hyperthermia. Mol Brain Res 75: 309-320.
- Brown IR (2007) Heat shock proteins and protection of the nervous system. Ann. N.Y. Acad. Sci 1113: 147-158.
- Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK (1999) Theneuroprotective potential of heat shock protein 70 (HSP70). Molecular Medicine Today 5: 525-531.
- Turturici G, Sconzo G, Geraci F (2011) Hsp70 and its molecular role in nervous system diseases. Biochem Res Int 2011: 1-18.
- Foster JA, Brown IR (1997) Differential induction of heat shock mRNA in oligodendrocytes, microglia, and astrocytes following hyperthermia. Brain Res Mol Brain Res 45: 207-218.
- Pavlik A, Aneja IS (2007) Cerebral neurons and glial cell types inducing heat shock protein Hsp70 following heat stress in the rat. Prog Brain Res 162: 417-431.
- Pavlik A, Aneja IS, Lexa J, Al-Zoabi BA (2003) Identification of cerebral neurons and glial cell types inducing heat shock protein Hsp70 following heat stress in the rat. Brain Res 973: 179-189.
- Pepper A, Grimbergen CA, Spaan JAE, Souren JEM, van Wijk R (1998) A mathematical model of the hsp70 regulation in the cell. Int. J. Hyperthermia 14: 97-124.
- Mohammadi S, Pavlik A, Krajci D, Al-sarraf H (2009) NMDA preconditioning and neuroprotection in vivo: delayed onset of kainic acid-induced neurodegeneration and c-Fos attenuation in CA3a neurons. Brain Res 1256: 162-172.
- Nadler JV, Perry BW, Cotman CW (1981) Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 29: 2031-2042.
- Olney JW (1978) Kainic acid as a tool in neurobiology. In: McGeer, E. G., Olney, J. W., and McGeer, P. L. (Eds). Neurotoxicity of Excitatory Amino Acids. Raven Press. New York pp. 95-121.
- Hatazaki S, Bellver-Estelles C, Jimenez-Mateos EM, Meller R, Bonner C, et al. (2007) Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neurosci 150: 467-477.
- Tang Y, Lu A, Aronow BJ, Wagner KR, Sharp FR (2002) Genomic responses of the brain to ischemic stroke, intracerebral hemorrhage, kainite seizures, hypoglycemia, and hypoxia. Eur J Neurosci 15: 1937-52.
- Zagulska-Szymczak S, Filipkowski RK, Kaczmarek L (2001) Kainate-induced genes in the hippocampus: lessons from expression patterns. Neurochem. Int 2001; 38: 483-501.
- Pringle AK, Thomas SJ, Signorelli F, Iannotti F (1999) Ischaemic preconditioning in organotypic hippocampal slice cultures is inversely correlated to the induction of the 72 kDa heat shock protein (HSP72). Brain Res 845: 152-164.
- Krueger AM, Armstrong JN, Plumier J, Robertson HA, Currie RW (1999) Cell-specific expression of Hsp70 in neurons and glia of the rat hippocampus after hyperthermia and kainic acid-induced seizure activity. Mol. Brain Res 71: 265-278.
- Brown IR (1994) Induction of heat shock genes in the mammalian brain by hyperthermia and tissue injury. In: Heat Shock Proteins in the Nervous System. (Eds) Mayer, R. J., and Brown, I. R. Academic Press, London 31-53.
- Duveau V, Arthaud S, Serre H, Rougier A, Salle GL (2005) Transient hyperthermia protects against subsequent seizures and epilepsy-induced cell damage in the rat. Neurobiol of Dis 19: 142-149.
- Duveau V, Arthaud S, Serre H, Rougier A, Salle GL (2007) Polysialylation of NCAM is upregulated by hyperthermia and participates in heat shock preconditioning- induced neuroprotection. Neurobiol of Dis 26: 385-395.
- Kourtis N, Nikoletopoulou V, Tavernaratis N (2012) Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 490: 213-218.
- Carruba MO, Bondiolotti G, Picotti GB, Catteruccia N, Da Prada M (1987) Effects of diethyl ether, halothane, ketamine and urethane on sympathetic activity in the rat. Eur J Pharmacol 134: 15-24.
- LeDuigou C, Wittner L, Danglot L, Miles R (2005) Effects of focal injection of kainic acid into the mouse hippocampus in vitro and ex vivo. Physiol 569: 833-847.
- Zhang SJ, Chen (1998) Spatial learning and memory induce up-regulation of nitric oxide producing neurons in rat brain. Brain Res 801: 101-106.
- Schmued LC, Albertson C, SlikkerJrW (1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res 751: 37-46.
- Sharp FR, Massa SM, Swanson RA (1999) Heat-shock protein protection. Trends Neurosci 22: 97-99.
- Chan RK, Sawchenko PE (1994) Spatially and temporally differentiated patterns of c-fos expression in brainstem catecholaminergic cell groups induced by cardiovascular challenges in the rat. J Comp Neurol 348: 433-460.
- Cobb JP, Karl RS, Karl IE, Buchman TG (1996) Mechanisms of cell injury and death. British Journal of Anesthesia 77: 3-10.
- Serova LI, Saez E, Spiegelman BM, Sabban EL (1998) c-Fos deficiency inhibits induction of mRNA for some, but not all, neurotransmitter biosynthetic enzymes by immobilization stress. J Neurochem 5: 1935-1940.
- Dragunow M, Faull R (1989) The use of c-fos as a metabolic marker in neuronal pathway tracing. J Neurosci Methods 29: 261-265.
- Morgan JI, Curran T (1991) Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu Rev Neurosci 14: 421-451.
- Sharma HS, Cervos-Navarro J, Dey PK (1991) Acute heat exposure causes cellular alteration in cerebral cortex of young rats. Neuroreport 2: 155-158.
- Monaghan DT, Cotman CW (1982) The distribution of [3H] kainic acid binding sites in rat CNS as determined by autoradiography. Brain Research 252: 91-100.
- Lee S, Sowa ME, Choi JM, Tsai FT (2004) The ClpB/Hsp104 molecular chaperone: a protein disaggregating machine. J. Struct. Biol 146: 99-105.
- Park HJ, Kim HJ, Park HJ, Ra J, Zheng LT, et al. (2008) Protective effect of topiramate on kainic acid-induced cell death in mice hippocampus. Epilepsia 49: 163-167.
- Hashimoto K, Watanabe KI, Iyo M, Shirayama Y, Minabe Y (1998) Behavioral changes and expression of heat shock protein hsp-70 mRNA in brain-derived neurotrophic factor mRNA, and cyclooxygenase-2-mRNA in rat brain following seizures induced by systemic administration of kainic acid. Brain Res 804: 212-223.
- Caputa M, Demicka A, Dokladny K, Kurowicka B (1996) Anatomical and physiological evidence for efficacious selective brain cooling in rats. J. Therm. Biol 21: 21-28.
- Sharma HS (2007) Heat shock proteins and the heat shock response during hyperthermia and its modulation by altered physiological conditions. Progress in Brain Research 162: 433-446.
- Xi L, Tekin D, Bhargava P, Kukreja RC (2001) Whole body hyperthermia and preconditioning of the heart: basic concepts, complexity, and potential mechanisms. Int J Hyperthermia 17: 439-455.
- Calderwood SK, Asea A (2002) Targeting HSP70-induced thermotolerance for design of thermal sensitizers. Int. J. Hyperthermia 18: 597-608.
- Lee YJ, Erdos, Hou ZZ, Kim SH, Kim JH, et al. (1994) Mechanism of quercetin-induced suppression and delay of heat shock gene expression and thermotolerance development in HT-29 cells. Mol Cell Biochem 137: 141-154.
- Wu BY, Yu AC (2000) Quercetin inhibits c-fos, heat shock protein, and glial fibrillary acidic protein expression in injured astrocytes. J Neurosci Res 62: 730-736.
- Jacubowitz-Gil J, Rzymowska J, Gawron A (2002) Quercetin, apoptosis, heat shock. BiochemPharmacol 2002; 64: 1591-1595.
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, et al. (2006) Small molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat ChemBiol 2: 474-479.
- Nijboer CH, Heijnen CJ, van der Kooij MA, Zijlstra J, van Velthoven CT, et al. (2011) Targeting the p53 pathway to protect the neonatal ischemic brain. Ann Neurol 2011; 70: 255-264.
- Tytell M, Greenberg SG, Lasek RJ (1986) Heat shock-like protein is transferred from glia to axon. Brain Res 363: 161-164.
- Tytell M (2005) Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int. J. Hyperthermia 21: 445-455.
- Karunanithi S, Barclay JW, Brown IR, Robertson RM, Atwood HL (2002) Enhancement of presynaptic performance in transgenic Drosophila overexpressing heat shock protein Hsp70. Synapse 44: 8-14.
- Kelty JD, Noseworthy PA, Feder ME, Robertson RM, Ramirez JM (2002) Thermal preconditioning and heat-shock protein 72 preserve synaptic transmission during thermal stress. J Neurosci 193: 1-6.
- Landry J, Bernier C, Chretien P, Nicole LM, Tanguay RM, et al. (1982) Synthesis and degradation of heat shock proteins during the development and decay of thermotolerance. Cancer Res 42: 2457-2461.
- Lindquist S, Craig EA (1988) The heat shock proteins. Annu Rev Genet 22: 631-677.
- El Bahh B, Auvergne R, Lere C, Brana C, Gal La Salle G, et al. (2001) Decreased epileptic susceptibility correlates with neuropeptide Y overexpression in a model of tolerance to excitotoxicity. Brain Res 894: 209-217.
- El Bahh B, Lurton D, Sundstrom LE, Rougier A (1997) Induction of tolerance and mossy fibre neuropeptide-Y expression in the contralateral hippocampus following a unilateral intrahippocampal KA injection in the rat. Neurosci. Lett 227: 135-139.

