Kallol Set*
Department of Pediatric Neurology, Children’s Hospital of Michigan, Detroit, Michigan 48201, USA
*Corresponding Author:
Kallol Set
MD, Department of Pediatric Neurology
Children’s Hospital of Michigan
Wayne State University, Detroit, MI 48201, USA
Tel: (1) 313-745-5788
E-mail: dr.kallolset@gmail.com
Received date: Sep 28, 2016; Accepted date: Sep 29, 2016; Published date: Sep 30, 2016
Citation: Set K. Images in Aicardi-Goutieres syndrome. Transl Biomed. 2016, 7:3. doi:: 10.21767/2172-0479.100088
Aicardi-Goutieres syndrome (AGS) is a very rare genetic condition. A 16-month old girl one of twin, was born from consanguineous marriage with microcephaly, global developmental delay, hypertonia, failure to thrive has been presented here. Extensive lab work and MRI images suggested differential diagnosis of Aicardi-Goutieres, megalencephalic leukoencephalopathy with subcortical cysts, CMV TORCH intrauterine infection. The molecular testing for Aicardi-Goutieres syndrome (AGS) was performed which revealed an apparent homozygous deletion of exons 14 and 15 in the SAMHD1 gene. These exons repeatedly failed to amplify for sequence analysis which is consistent with a diagnosis of Aicardi-Goutieres syndrome. Symptomatic management and Genetic counselling done.
Introduction
Aicardi-Goutieres syndrome (AGS) manifests as an earlyonset encephalopathy with severe intellectual and physical handicap after the first few weeks of life, frequently after a period of apparently normal development [1-3]. Typically, they present with sub-acute onset of a severe encephalopathy characterized by extreme irritability, intermittent sterile pyrexias, loss of skills, and slowing of head growth. Over time, as many as 40% develop chilblain skin lesions on the fingers, toes, and ears [3]. I present a patient with all the above mentioned features.
A 16-month old girl, born of consanguineous marriage with microcephaly, global developmental delay, hypertonia, failure to thrive, extreme irritability, intermittent fever. Lab work shows Normal urine organic acids, normal plasma amino acids analysis, normal liver function testing, normal acylcarnitine profile, a plasma lactate with mildly elevated. Spinal tap with CSF neopterin (elevated at greater than 300 nmol/L), reference range 7-65. CGH array showed a 2.31 Mb microdeletion at Xp22.11p21.3, containing 15 genes including the ARX gene. MRI of brain explained in images (Figure 1). Differential diagnosis was Acardi-Goutieres (given the microcephaly, calcifications, periventricular white matter changes, and large cisterna magna and midline cyst, however there is no definite polymicrogyria or ocular coloboma), megaloencephalic Leukoencephalopathy with subcortical cysts (given the extensive white matter and cystic changes, however there is microcephaly and not macrocephaly), prior CMV TORCH infection (given the calcifications, however it is less likely given that the other twin was asymptomatic). Had extensive workup including TORCH titers, PCR for CMV and urine CMV studies, all been negative. The molecular testing for AGS was performed at Denver genetic laboratories and including the TREX1, RNASEH2A, RNASEH2B, RNASEH2C and SAMHD1, revealed an apparent homozygous deletion of exons 14 and 15 in the SAMHD1 gene. these exons repeatedly failed to amplify for sequence analysis which is consistent with a diagnosis AGS [1]. Mother apparently is a carrier for AGS and father does have the mutation again in SAMHD1 though he is asymptomatic. Genetic counselling and symptomatic management done.
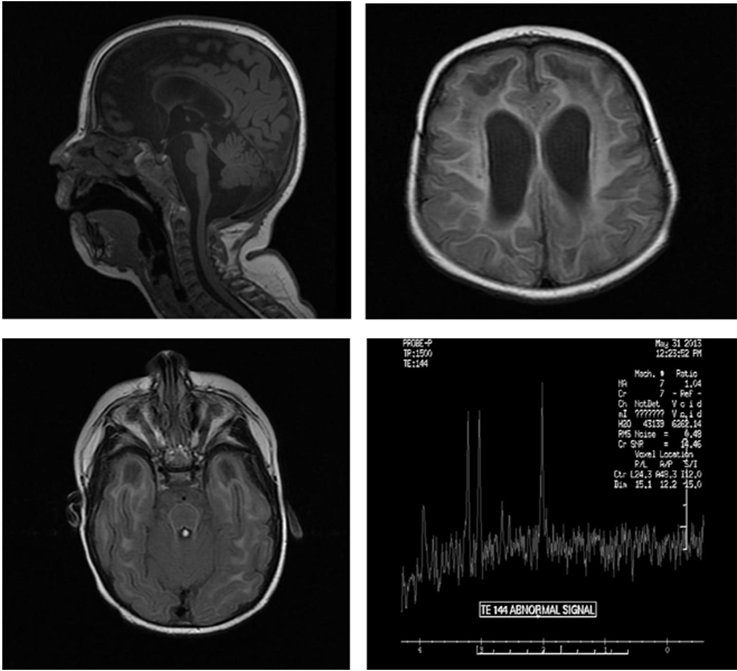
Figure 1: MRI of Brain shows microcephaly, moderate ventriculomegaly, diffuse decreased caliber of the brainstem, thin corpus callosum. Increased amount CSF signal in the posterior fossa likely represents a prominent cisterna magna. Extensive abnormal hyper intense T2/FLAIR signal seen throughout the periventricular and subcortical white matter of the frontal, parietal, temporal and occipital lobes. There are subcortical cystic changes of bilateral anterior temporal lobes and anterior right frontal lobe. MR spectroscopy with voxel positioning in the left frontal lobe demonstrates decreased choline, creatinine and NAA on the TE 38 and 144 signal. Evaluation of the lactate peak is difficult due to artifact. Overall nonspecific.
17230
References
- Aicardi J, Goutières F (1984) A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15: 49-54.
- Millichap JG (2015) MRI Features Predictive of Aicardi-Goutieres Syndrome. Ped Neurol Briefs 29: 8.

