Keywords
Casein kinase 2; CK2; Flavonoids; Breast cancer; Molecular docking; Pharmacophore modelling; Structure based drug design
Introduction
Protein kinase II, also known as casein kinase II (CK2), is a constitutively active, ubiquitously expressed serine/threonine protein kinase. This protein exists as a heterotetrameric complex and is composed of an alpha, an alpha-prime, and two beta subunits. The alpha subunits contain the catalytic activity while the beta subunits undergo autophosphorylation. It has demonstrated that CK2 is involved in a wide variety of cellular processes such as gene expression, protein synthesis, proliferation, apoptosis and differentiation/ transformation [1-6]. Thus, perturbations in expression or activity of CK2 are associated with human disease [7-10]. Recently, it has demonstrated that this protein is involved in several human cancers, including cancer of the breast, lung, colon, and prostate, as well as hematologic malignancies [11,12]. In the light of these observations, CK2 represents an attractive pharmacological target for the treatment of cancer and the development of specific inhibitors of CK2. Therefore, the development and design of more promising CK2 inhibitors would be highly desirable.
A huge number of CK2 inhibitors are available, most of them are able to directly target the adenosine triphosphate (ATP) binding site, such as 5,6-Dichloro-1-β-D-ribofuranosyl-benzimidazole (DRB), emodin, tetrabromobenzotriazole (TBB), apigenin (AGI), and [5- oxo-5,6- dihydroindolo(1,2-a) quinazolin-7-yl] acetic acid (IQA) [13,14]. AGI is a flavonoid compound, belonging to a group of natural substances, which effectively inhibits CK2. This inhibitor, like most protein kinase inhibitors, is competitive with respect to the ATP. This indicates that AGI is directed towards the conserved ATP binding site [15]. However, AGI is more potent, but as other general flavonoids, its specificity is quite broad [14,16]. Thus, there is a need to design new compounds which are able to inhibit CK2 with selectivity higher than those of AGI.
The aim of this investigation is to identify new CK2 inhibitors. In this context, we are interested in the ligand-protein binding, which is important not only as an essential molecular recognition process but also for the discovery of new CK2 inhibitors, between a series of flavonoids derivatives and CK2 protein. Molecular docking is an in-silico approach that is used to predict the best binding mode of compound within the active site of the specified enzyme. This technique has been applied previously in drug discovery [17-19] and many molecular modeling softwares are available among them the FlexX program [20].
In the present study, we performed a molecular docking study to search for best binding mode of a series of thirty-seven hydroxyflavones derivatives in the CK2 active site by using FlexX program. The obtained conformations of some active compounds are used to generate a pharmacophore model which can be applied to drug design and virtual screening [21,22].
Materials and Methods
Molecular docking
Ligands preparation: The dataset used in this study, characterized by adequate biological and structural diversity, belongs to flavonoids family (Figure 1). Thirty-seven hydroxy flavones derivatives, which exhibited experimental activities ranged from 0.012 to 30 μM, were selected as ligands [23]. These activities, are given in terms of IC50, the drug concentration that inhibits 50% of the enzymatic activity of CK2, which were converted to pIC50. The two-dimensional structure of these compounds (Table 1) were drawn with ISIS 2.4 package [24]. The obtained geometries were energy optimized using the steepest descent approximation and were converged to a gradient of 0.02 using the UCSF Chimera software [25]. Table 1 provides the negative logarithms of the observed activities, denoted PIC50.

Figure 1: General structure of flavonoids derivatives studied in this work.
| ID |
R1’ |
R2’ |
R3’ |
R4’ |
R4 |
R5 |
R6 |
R7 |
R8 |
P IC50 |
| F1 |
H |
NO2 |
OH |
H |
H |
H |
Me |
H |
Me |
-1.92 |
| F2 |
H |
OMe |
OH |
H |
H |
H |
H |
C4H4 |
---- |
-1.55 |
| F3 |
H |
Br |
OH |
H |
H |
H |
Me |
H |
Me |
-1.30 |
| F4 |
H |
OMe |
OH |
NO2 |
H |
H |
Me |
H |
Me |
-1.18 |
| F5 |
H |
OMe |
OH |
Cl |
H |
H |
H |
C4H4 |
H |
-1.06 |
| F6 |
H |
OMe |
OH |
H |
H |
H |
H |
Me |
H |
-1.05 |
| F7 |
H |
OMe |
OH |
H |
H |
H |
Me |
H |
Me |
-1.00 |
| F8 |
H |
OMe |
OH |
H |
H |
H |
Et |
H |
H |
-0.92 |
| F9 |
H |
OMe |
OH |
H |
H |
H |
Br |
H |
H |
-0.92 |
| F10 |
H |
OMe |
OH |
H |
H |
H |
NHAc |
H |
H |
-0.82 |
| F11 |
H |
OMe |
OH |
H |
H |
H |
OMe |
H |
H |
-0.76 |
| F12 |
H |
OMe |
OH |
H |
H |
H |
OMe |
H |
H |
-0.70 |
| F13 |
H |
OMe |
OH |
H |
H |
H |
Cl |
H |
H |
-0.62 |
| F14 |
H |
OMe |
OH |
H |
H |
H |
OH |
H |
H |
-0.60 |
| F15 |
H |
OMe |
OH |
H |
H |
H |
H |
Me |
Me |
-0.52 |
| F16 |
H |
OMe |
OH |
H |
H |
H |
Me |
H |
H |
-0.49 |
| F17 |
H |
OH |
OH |
H |
H |
H |
Me |
H |
Me |
-0.35 |
| F18 |
H |
OMe |
OH |
H |
H |
H |
Me |
H |
Me |
-0.19 |
| F19 |
H |
OMe |
OH |
H |
H |
H |
Cl |
Me |
H |
-0.10 |
| F20 |
H |
OMe |
OH |
H |
H |
H |
H |
H |
H |
-0.10 |
| F21 |
H |
H |
OH |
H |
H |
H |
Me |
H |
Me |
+0.20 |
| F22 |
H |
OMe |
OH |
H |
H |
Me |
Cl |
Me |
H |
+0.30 |
| F23 |
H |
OH |
H |
H |
H |
H |
Me |
H |
Me |
+0.34 |
| F24 |
H |
OMe |
OH |
H |
H |
H |
Me |
Me |
H |
+0.48 |
| F25 |
H |
OEt |
OH |
H |
H |
H |
H |
H |
H |
+0.88 |
| F26 |
H |
OEt |
OH |
H |
H |
H |
Me |
H |
H |
+0.90 |
| F27 |
H |
H |
OH |
H |
H |
H |
Me |
H |
H |
+0.90 |
| F28 |
H |
H |
Me |
H |
H |
H |
OH |
H |
H |
+1.28 |
| F29 |
H |
Br |
H |
H |
H |
H |
OH |
H |
H |
+1.28 |
| F30 |
H |
H |
Me |
H |
H |
H |
Me |
H |
Me |
+1.32 |
| F31 |
H |
H |
Cl |
H |
OH |
H |
Me |
H |
H |
+1.36 |
| F32 |
H |
OMe |
H |
H |
OH |
H |
Me |
H |
H |
+1.40 |
| F33 |
H |
H |
i-Pr |
H |
OH |
H |
Me |
H |
H |
+1.41 |
| F34 |
H |
OMe |
OMe |
OMe |
H |
H |
OH |
H |
H |
+1.41 |
| F35 |
OH |
H |
H |
Br |
OH |
H |
Me |
H |
H |
+1.45 |
| F36 |
H |
H |
OMe |
H |
OH |
H |
Me |
H |
H |
+1.45 |
| F37 |
H |
H |
OMe |
H |
OSO2Me |
H |
Me |
H |
Me |
+1.45 |
Table 1: Structures and PIC50 of thirty seven flavonoids derivatives.
Receptor preparation: There are many CK2 complex crystal structures available in Protein Data Bank [26]. Based on the resolution, the crystal structure of CK2 bound with AGI (Apigenin) (code 4DGM determined at 1.65 Å of resolution) was selected as a receptor [27]. The protein was prepared using Receptor Preparation Wizard of LeadIT software from BioSolveIT, GmbH Germany (version 2.2.0) [28] by adding polar hydrogen.
Binding site determination: In LeadIT software, the selection of the binding site can be performed using two methods; the reference ligand and the selection of sphere to get the best score. In this work, the active site of the CK2 protein was determined based on the cocrystallized ligand (AGI) in the crystal structure. The active site radius was taken to be 6.5 Å from the center of co-crystallized ligand at the site of docking; so that the amino acid residues namely Val116, Glu114, Ile66, Met163, Val95, Val53, Val45 and Ile174 were included.
Docking protocol: The docking of flavonoids derivatives in the CK2 active site was performed using the FlexX docking algorithm [20] implemented in LeadIT program. The SDF file of all compounds was loaded in FlexX as a docking library. Then, the compounds were docked in the CK2 active site using default docking parameters and 50 highest scoring docked solutions of each compound were selected for further analysis. The interactions present in the selected dock poses were represented in 2D format using protein-ligand interaction map generating widget, PoseView [29] of LeadIT program. In order to validate obtained docking results, the correlation between the docking score values in Kcal/mol and the inhibitory activity of the selected compounds was computed.
Binding free energy calculations: FlexX is one of the best suited software for the analysis of macromolecule-ligand Interactions [30]. The docking algorithm in FlexX uses flexible ligand and rigid protein [31]. Ligand flexibility limited to torsion angles and ring conformations. This algorithm is based on the model of molecular interactions defined by Böhm [32,33]. This model can be divided into three areas: conformational flexibility, protein-ligand interactions, and the scoring function [20,34]. The ranking of the generated solutions is performed using a Böhm scoring function which estimates the free binding energy (ΔGbinding) of the protein-ligand complex [33-35]. ΔGbinding can be calculated using the equation 1 (Eq.1). ΔG0 corresponds to the loss of translational and rotational entropy of the ligand upon binding. ΔGpolar and ΔGapolar correspond to polar and a polar interaction, respectively. ΔGsolv and ΔGflexi are energies of solvation and flexibility of the ligand structure.

Structure-Based Pharmacophore Generation
In the present study, the structure-based pharmacophore model was generated using the LigandScout software package [36] to identify the interaction of critical residues between the protein and ligand. LigandScout software uses an algorithm that studies and interprets ligand-receptor interactions and then automatically creates an advanced pharmacophore model. As input for structure-based pharmacophore generation, the obtained docked complexes of some active compounds with CK2 protein were chosen. Following this, a new structure based pharmacophore was generated by selecting the most relevant structural features of these compounds and removing the overlapped chemical features.
Results and Discussion
Comparison between the docked and X-ray crystal structures
The docked conformation of AGI was close to the crystal structure (Figure 2), since the RMSD between the two conformations was only 0.75 Å, which is quite satisfactory. This suggests that the FlexX docking method is reasonable and reliable. Therefore, the studied compounds can be docked correctly to the CK2 active site.

Figure 2: a-Comparison between the X-ray structure and the docking structure for AGI (sticks: docking, ball and sticks: crystal structure). b- Docked conformation of AGI in the CK2 active site.
Docking interaction analysis
Molecular docking was carried out to interpret the best binding pose of the ligands and search the similar chemical entities that can bind strongly with the active site residues of CK2 protein. Based on the obtained docking results, ligands bind at the same ATP-binding site of CK2 protein as determined experimentally (Figure 3). Furthermore, the ligands poses are stabilized by a combination of hydrogen bonds and hydrophobic interactions with active site residues. Analysis of results revealed that water molecule Hoh581 and Val116 were the common residues in most interactions between carbonyl group (CO) of studied compounds and CK2 protein. Asp175, Lys68, Val45, Asn118, Arg47, Val116, Glu114 and Asp120 were the main amino acid residues involved in hydrogen bonding with the compounds which exhibited low activities (more potent compounds) toward CK2 (Figure 4). Nitro group (NO2) of compounds F1 and F4 was very active; it favors the hydrogen bond interactions. This group is pointed toward the positive charge region that is located near the amine function (NH3) of the Lys68 side chain. It interacts with protein CK2 forming hydrogen bonds with backbone nitrogen of Asp175 and amino group (NH3) of Lys68. This result is in good agreement with previous studies in which the authors demonstrate that the ligands that carry the negatively charged moiety adopt the conformation in which the negative group is pointed toward the positively charged region of the ATP binding site located in the buried area of the cavity (near the well conserved salt bridge Lys68- Glu81) [37]. This region is responsible for polar interactions with the negatively charged moiety of the ligand, contributing to the different orientation of the ligand itself [37].

Figure 3: View of the most favorable conformation of each compound (shown as a stick) docked into the CK2 active site.
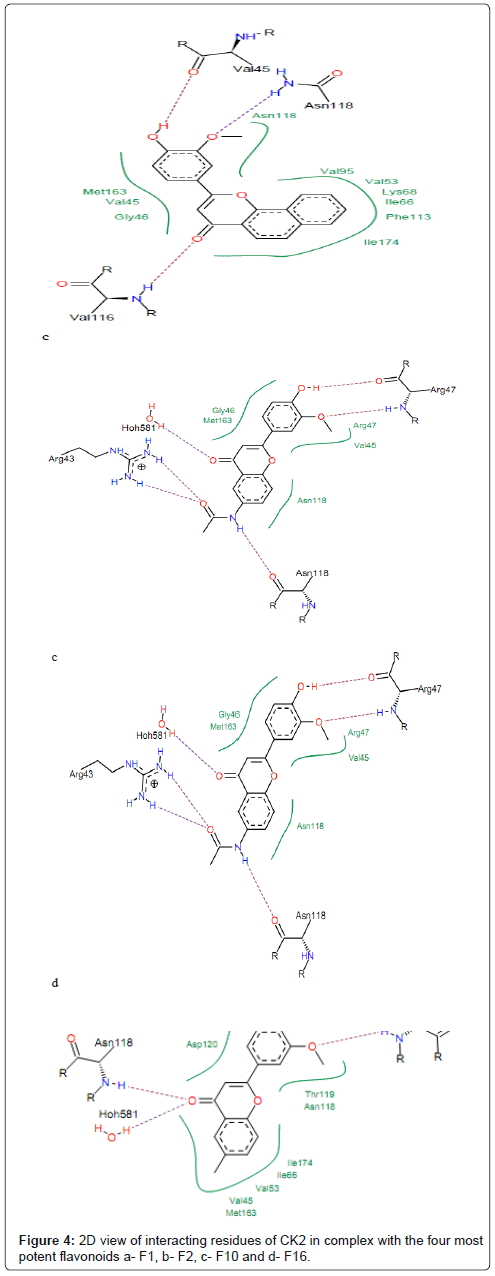
Figure 4: 2D view of interacting residues of CK2 in complex with the four most potent flavonoids a- F1, b- F2, c- F10 and d- F16.
The hydroxyl oxygen at position 3’ (Figure 1) of compounds F2, F5, F13, F16 and F20 is hydrogen bonded to the backbone nitrogen of Asp120 and Arg47. The compounds F17, F16, F18 and F19 interact with protein forming hydrogen bond between hydroxyl group (-OH) and residue Asp120 (Figure 4). The backbone oxygen and backbone nitrogen of Arg47 showed strong hydrogen bonds with the methoxy group (-OMe) in position 2’ and hydroxyl oxygen in position 3’ of compounds F10, F17, F16, F18, F19, respectively. The residues Asn118, Gly46, Met163, Val45, Phe113, Val95, Val53, Ile174 and Ile66 made hydrophobic interactions with these compounds (Figure 4). The aromatic rings A of compounds F1, F4 and F16 are embedded into hydrophobic cavity consisting of Asn118, Gly46, Met163 and Val45. This indicates that all these compounds fit well in the binding pocket of CK2 protein which may explain their highest affinities toward CK2. It is worth noting that, for many of the studied compounds, hydrophobic interactions and hydrogen bonds are the most important driving force that stabilizes their binding to CK2 and, hence, are mainly responsible for the potency of the inhibitor [38]. However, we judged the binding mode proposed by FlexX for these compounds as quite plausible.
The compounds which exhibited the PIC50 ranging from 0 to 1 (050<1), interact with protein forming hydrogen bonds between their hydroxyl oxygen at 3’ position (2’ for compound F23) and residues Asp120 (Figure 5). The visualization of docked conformation of compound F22 shows that Asp120 had two hydrogen bonds, one with hydroxyl oxygen (–OH) in position 3’ and one other with methoxy group (–OCH3) at position 2’ (Figure 5). This allows multiple hydrophobic interactions to be made between the polycyclic core of the molecule and residues Asp120, Val53, Met163, Ile66, Val45, Ile174, Thr119 and Asn118 that normally form the environment of the adenosine part of ATP in the enzyme pocket. From Figure 3, we can see that the aromatic rings A of compounds F22 and F24 are surrounded by Ile66, Val45, Ile174, Val53 and Met163.
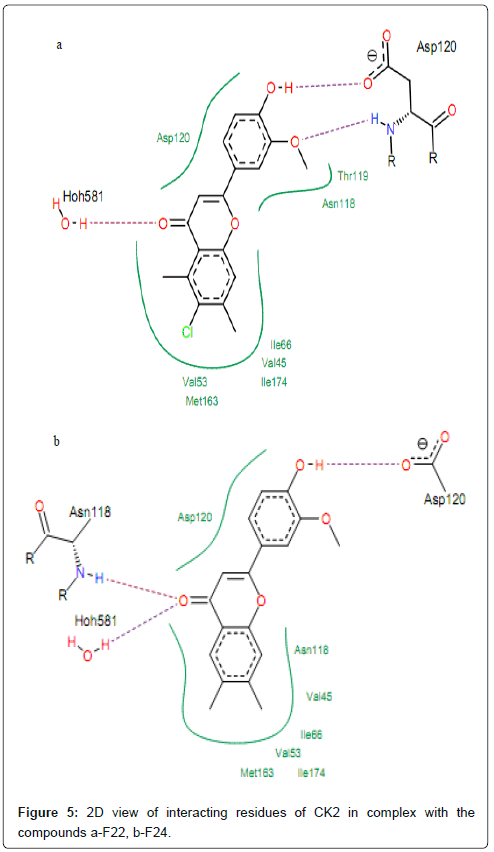
Figure 5: 2D view of interacting residues of CK2 in complex with the compounds a-F22, b-F24.
H-bonds formed by interaction of compounds with high activities (PIC50>1) and CK2 residues Val116 and Hoh581. The hydroxyl oxygen (-OH) at position R8 of compound F34 is involved in hydrogen bonding with backbone nitrogen of Val116 and backbone oxygen of Glu114. This group at position 3 of compounds F35 and F36 is hydrogen bonded with backbones oxygen and nitrogen of residue Val116 (same interactions are observed for compounds F31, F32 and F33), as shown in Figure 6. It is worth noting that, for some CK2 inhibitors which exhibited high activities, the preferred interaction is with the backbone carbonyls of Glu114 and Val116. Other previous docking studies of CK2 inhibitors also discovered according results [38]. Moreover, Sarno et al. [36] clarified that backbone of residues Glu114 and Val116 was the key amino acid residue inside the binding site of CK2 responsible for the interaction with pentabromoindazole (K64). Residues Met163 is the common residue linked to the ring A of compounds F34, F35 and F36 via hydrophobic interactions.
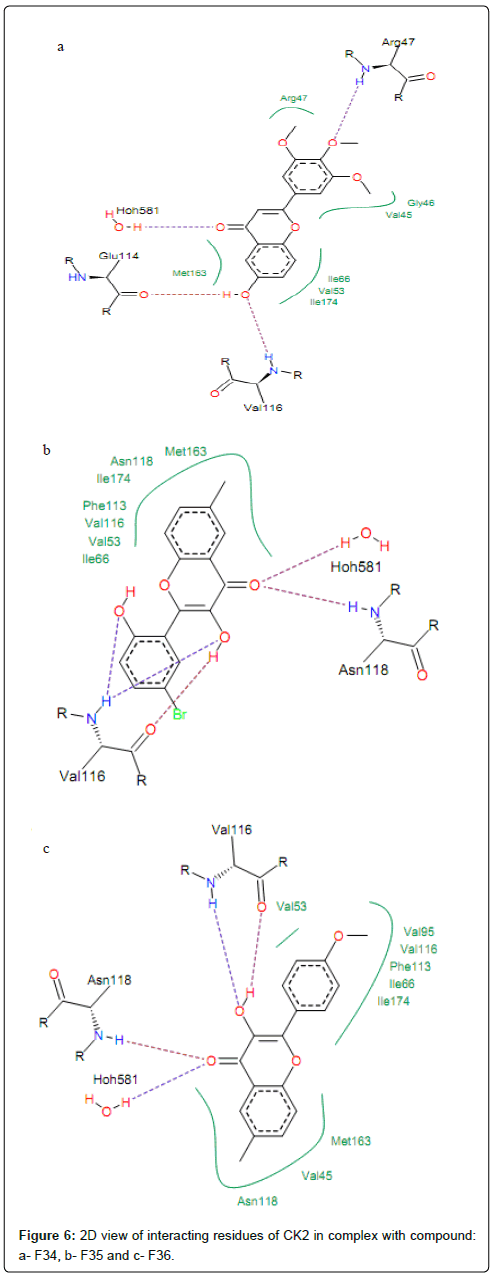
Figure 6: 2D view of interacting residues of CK2 in complex with compound: a- F34, b- F35 and c- F36.
Docking and experimental energies of selected compounds: The predicted docking energies score, obtained by FlexX program for studied compounds and the corresponding experimental PIC50 values are listed in Table 2. More negative values pointed out more favourable binding.
The PIC50 values of ligands can be grouped into three classes, active, inactive or moderately active, based on whether the PIC50 value is less than -1, more than 1 or ranging from -1 to 1, respectively. The performance of docking models is mostly measured in terms of their ability to discriminate between active and inactive compounds by calculating the docking energies. Thus, the actives and inactive compounds must be classified in active and inactive classes, respectively. Analysis of data presented in the Table 2 shows that compounds F1, F2, F4, F5 and F6 are well classified in the active class and compounds F2- F35 and F37 are well classified in the inactive class. Thus, we can say that the active molecules are 72% correctly classified, while the inactive molecules are 80% correctly classified. The correlation between the calculated energies score values of the studied compounds and the experimental PIC50 values is studied and plotted in Figure 7, which displays a good linear relationship. It is obvious from this figure that all the docking energies are in good agreement with the corresponding experimental activities since a significant correlation coefficient R=0.866 (R2=0.750) was obtained.
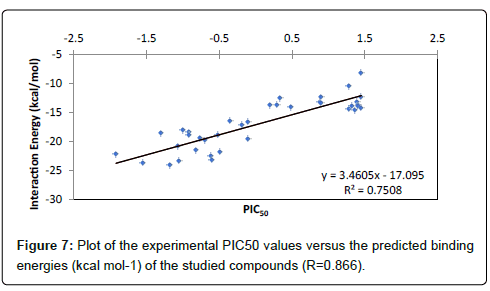
Figure 7: Plot of the experimental PIC50 values versus the predicted binding energies (kcal mol-1) of the studied compounds (R=0.866).
| mol |
PIC50 |
Score |
| F1 |
-1.92 |
-22.19 |
| F2 |
-1.55 |
-23.64 |
| F3 |
-1.30 |
-18.56 |
| F4 |
-1.18 |
-24.09 |
| F5 |
-1.06 |
-20.86 |
| F6 |
-1.05 |
-23.34 |
| F7 |
-1.00 |
-18.08 |
| F8 |
-0.92 |
-18.44 |
| F9 |
-0.92 |
-18.87 |
| F10 |
-0.82 |
-21.50 |
| F11 |
-0.76 |
-19.47 |
| F12 |
-0.70 |
-19.83 |
| F13 |
-0.62 |
-22.44 |
| F14 |
-0.60 |
-23.11 |
| F15 |
-0.52 |
-18.85 |
| F16 |
-0.49 |
-21.80 |
| F17 |
-0.35 |
-16.47 |
| F18 |
-0.19 |
-17.08 |
| F19 |
-0.10 |
-19.5 |
| F20 |
-0.10 |
-16.59 |
| F21 |
+0.20 |
-13.76 |
| F22 |
+0.30 |
-13.67 |
| F23 |
+0.34 |
-12.59 |
| F24 |
+0.48 |
-14.08 |
| F25 |
+0.88 |
-13.19 |
| F26 |
+0.90 |
-13.45 |
| F27 |
+0.90 |
-12.38 |
| F28 |
+1.28 |
-14.38 |
| F29 |
+1.28 |
-10.50 |
| F30 |
+1.32 |
-13.88 |
| F31 |
+1.36 |
-14.67 |
| F32 |
+1.40 |
-13.17 |
| F33 |
+1.41 |
-13.75 |
| F34 |
+1.41 |
-13.87 |
| F35 |
+1.45 |
-12.30 |
| F36 |
+1.45 |
-8.18 |
| F37 |
+1.45 |
-14.23 |
Table 2: Docking energies score (kcal mol-1) and PIC50 values of the studied compounds.
Structure based pharmacophore modelling
Obtained docking results were imported in LigandScout. Molecule and side chains of each obtained complex were energy minimized using MMFF94 to refine the FlexX output. Then, important molecular interactions to binding site residues of CK2 protein such as hydrogen bonds, hydrophobic interactions, and so on were analyzed in order to identify important features for ligand binding. The obtained pharmacophore models show interactions between important amino acid residues in the binding site and ligand and then elucidate the favorable binding position of the docked compounds. Pharmacophore features in the generated models consist of hydrogen bond donor HBD (green arrow), hydrogen bond acceptor HBA (red arrow) and hydrophobic interaction Hy (yellow sphere); as shown in Figure 8 and listed in Table 3.
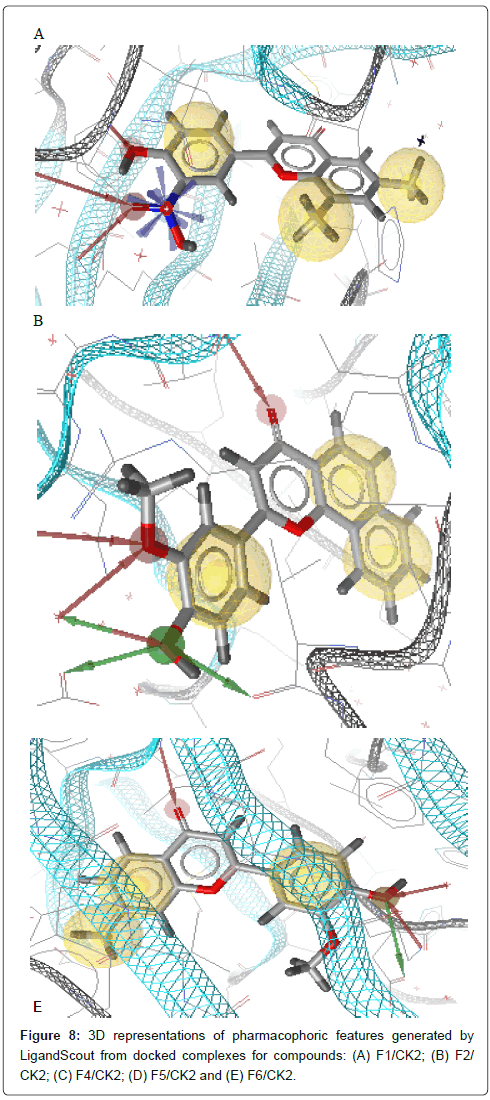
Figure 8: 3D representations of pharmacophoric features generated by LigandScout from docked complexes for compounds: (A) F1/CK2; (B) F2/ CK2; (C) F4/CK2; (D) F5/CK2 and (E) F6/CK2.
| mol |
R2’ |
R3’ |
R4’ |
|
R6 |
R7 |
R8 |
Ring A |
Ring C |
| F1 |
HBA+Pos ioni |
HBA |
|
|
Hy |
|
Hy |
|
Hy |
| F2 |
|
HBA+HBD |
HBA |
HBA |
|
|
|
Hy |
Hy |
| F4 |
HBA+Pos ioni+HBD |
HBA+HBD |
|
|
Hy |
|
Hy |
|
Hy |
| F5 |
|
HBD |
Hy |
HBA |
|
|
|
Hy |
|
| F6 |
|
HBA+HBD |
|
HBA |
|
Hy |
|
Hy |
Hy |
Table 3: Pharmacophoric features of each well classified compound in the active class.
The final pharmacophore features at each position in the common structure of the five well classified compounds, in the active class (compounds F1, F2, F4, F5 and F6) (Figure 8), were analyzed and clustered according to their interaction pattern with the CK2 protein. The most relevant structural features of these compounds were selected and included in the final pharmacophore model.
The analysis of the results presented in Table 3 shows that HBA and HBD in 3’ position is present in almost all compounds (four out of five). Three compounds out of five make an HBA between carbonyl group (CO) and CK2 active site residues. Two hydrophobic interactions between two rings A and C and CK2 protein should be taken into consideration. Hense, the final pharmacophore model consists of two HBA at positions 3’ and 4, one HBD at position 3’ and two hydrophobic interactions centers, ring A and ring C (Figure 9).
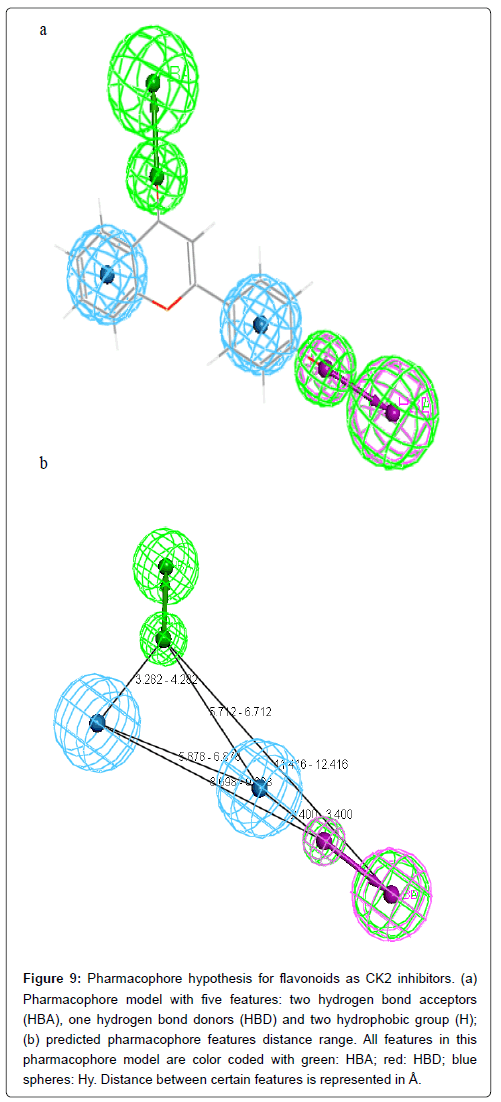
Figure 9: Pharmacophore hypothesis for flavonoids as CK2 inhibitors. (a) Pharmacophore model with five features: two hydrogen bond acceptors (HBA), one hydrogen bond donors (HBD) and two hydrophobic group (H); (b) predicted pharmacophore features distance range. All features in this pharmacophore model are color coded with green: HBA; red: HBD; blue spheres: Hy. Distance between certain features is represented in Å.
Conclusion
Thirty-seven flavonoids derivatives were docked on CK2 active site using the FlexX software. The docked conformation of the apigenin, extracted from the crystallographic complex 4DGM, was close to the crystal structure. Visualization of docking results shows that flavonoids derivatives interact with CK2 protein predominantly through hydrogen bonding. The majority of studied compounds were observed to form hydrogen bonds between either the carbonyl group (CO) of each compound and Val116 of the CK2 and with water molecule Hoh581 or between the hydroxyl oxygen and Asp120 and Arg47. The residues Asn118, Asp120, Ile66, Met163 and Val45 are linked to the ligands via hydrophobic interactions with the phenyl rings A and C. In order to add robustness to the employed docking model, the correlation between the docking energies of studied compounds and experimental PIC50 values was studied and plotted. The results show that the docked energies are in good agreement with the corresponding experimental activities since a significant correlation coefficient (R=0.866) was obtained.
Obtained docking conformations of the well classified active compounds were used to generate pharmacophore models with the help of LigandScout. the final pharmacophore model consists of two HBA at positions 3’ and 4, one HBD at position 3’ and two hydrophobic interaction at ring A and ring C. Therefore, the information provided in this study can be useful for designing new CK2 based on flavonoids compounds.
Acknowledgements
The authors would like to thank Inte:Ligand Software-Entwicklungs und Consulting GmbH and BioSolveIT team for providing an academic free license for LigandScout 4.1.7 and LeadIT 2.2.0 programs.
References
- Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369: 1-15.
- Filhol O, Martiel JL, Cochet C (2004) Protein kinase CK2: a new view of an old molecular complex. EMBO Rep 5: 351-355.
- Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17: 349-368.
- Pinna LA (2002) Protein kinase CK2: a challenge to canons. J Cell Sci 115: 3873-3878.
- Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2: a key suppressor of apoptosis. Adv Enzyme Regul 48: 179-187.
- Raaf J, Brunstein E, Issinger OG, Niefind K (2008) The interaction of CK2αand CK2β, the subunits of protein kinase CK2, requires CK2βin a preformed conformation and is enthalpically driven. Protein Sci 17: 2180-2186.
- Lin JM, Kilman VL, Keegan K, Paddock B, Emery-Le M, et al. (2002) A role for casein kinase 2 alpha in the Drosophila circadian clock. Nature 420: 816-820.
- Akten B, Jauch E, Genova GK, Kim EY, Edery I, et al. (2003) A role for CK2 in the Drosophila circadian oscillator. Nat Neurosci 6: 251-257.
- Blau J (2003) A new role for an old kinase: CK2 and the circadian clock. Nat Neurosci 6: 208-210.
- Daniel X, Sugano S, Tobin EM (2004) CK2 phosphorylation of CCA1 is necessary for its circadian oscillator function in Arabidopsis. Proc Natl Acad Sci USA 101: 3292-3297.
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355-365.
- Brognard J, Hunter T (2011) Protein kinase signaling networks in cancer. Curr Opin Genet Dev 21: 4-11.
- Zandomeni R, Zandomeni RC, Shugar D, Weinmann R (1986) Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem 261: 3414-3420.
- Sarno S, De Moliner E, Ruzzene M, Pagano MA, Battistutta R, et al. (2003) Biochemical and 3D-structural data on the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a) quinazolin-7-yl] acetic acid (IQA). Biochem J 374: 639-646.
- Sarno S, Salvi M, Battistutta R, Zanotti G, Pinna LA (2005) Features and potentials of ATP-site directed CK2 inhibitors. Biochim Biophys Acta 1754: 263-270.
- Agullo G, Gamet-Payrastre L, Manenti S, Viala C, Rémésy C, et al. (1997) Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition, Biochem Pharmacol 53: 1649-1657.
- Min L, Shuying M, Yueli T, Xiaoyun Z, Honglin Z, et al. (2015) Structural insights into avones as protein kinase CK2 inhibitors derived from a combined computational study. RSC Adv 5: 462.
- Souhila BT, Amel TM, Boubekeur M, Safia TK (2010) Modeling the binding modes of stilbene analogs to cyclooxygenase-2: a molecular docking study. J Mol Model 16: 1919-1929.
- Souhila BT, Redouane T, SaÂa TK (2013) Receptor and ligand-based 3D-QSAR study on a series of nonsteroidal anti-in¡ammatory drugs. Med Chem Res 22: 1529-1537.
- Rarey M, Kramer B, Lengauer T, Klebe G (1996) A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261: 470-489.
- Wolber G, Seidel T, Bendix F, Langer T (2008) Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today 13: 23-29.
- Yang SY (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 15: 444-450.
- Golub AG, Bdzhola VG, Ostrynska OV, Kyshenia IV, Sapelkin VM, et al. (20130) Discovery and characterization of synthetic 4’-hydroxyflavones-New CK2 inhibitors from flavone family. Bioorganic & Medicinal Chemistry 21: 6681-6689.
- MDL Information Systems (2002) ISIS/Draw 2.5. MDL Information System, San Leandro.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) USCF Chimera - A visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612.
- A Structural View of Biology. RCSB, Protein National Bank.
- Lolli G, Cozza G, Mazzorana M, Tibaldi E, Cesaro L, et al. (2012) Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight Biochemistry 51: 6097-6107.
- Lead IT (2014) Available from: https://www.biosolveit.de/LeadIT/
- Stierand K, Rarey M (2007) From modeling to medicinal chemistry: automatic generation of two-dimensional complex diagrams. Chem Med Chem 2: 853-860.
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, et al. (2006) A Critical Assessment of Docking Programs and Scoring Functions. J Med Chem 49: 5912-5931.
- Mukesh B, Rakesh K (2011) Molecular Docking: a Review. International Journal of Research in Ayurveda and Pharmacy 2: 1746-1751.
- Böhm HJ (1992) The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des 6: 61-78.
- Böhm HJ (1994) The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J Comput Aided Mol Des 8: 243-256.
- Rarey M, Kramer B, Lengauer T (1997) Multiple automatic base selection: protein-ligand docking based on incremental construction without manual intervention. J Comput Aided Mol Des 11: 369-384.
- Böhm HJ (1998) Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput-Aided Mol Des 12: 309-309.
- Wolber G, Langer T (2005) LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 45: 160-169.
- Battistutta R, Mazzorana M, Cendron L, Bortolato A, Sarno S, et al. The ATP-Binding Site of Protein Kinase CK2 Holds a Positive Electrostatic Area and Conserved Water Molecules. Chem Bio Chem 8: 1804-1809.
- Sarno S, Papinutto E, Franchin C, Bain J, Elliott M, et al. (2011) ATP Site-Directed Inhibitors of Protein Kinase CK2: An Update. Curr Top Med Chem 11: 1340-1351.
20414
References
- Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369: 1-15.
- Filhol O, Martiel JL, Cochet C (2004) Protein kinase CK2: a new view of an old molecular complex. EMBO Rep 5: 351-355.
- Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17: 349-368.
- Pinna LA (2002) Protein kinase CK2: a challenge to canons. J Cell Sci 115: 3873-3878.
- Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2: a key suppressor of apoptosis. Adv Enzyme Regul 48: 179-187.
- Raaf J, Brunstein E, Issinger OG, Niefind K (2008) The interaction of CK2αand CK2β, the subunits of protein kinase CK2, requires CK2βin a preformed conformation and is enthalpically driven. Protein Sci 17: 2180-2186.
- Lin JM, Kilman VL, Keegan K, Paddock B, Emery-Le M, et al. (2002) A role for casein kinase 2 alpha in the Drosophila circadian clock. Nature 420: 816-820.
- Akten B, Jauch E, Genova GK, Kim EY, Edery I, et al. (2003) A role for CK2 in the Drosophila circadian oscillator. Nat Neurosci 6: 251-257.
- Blau J (2003) A new role for an old kinase: CK2 and the circadian clock. Nat Neurosci 6: 208-210.
- Daniel X, Sugano S, Tobin EM (2004) CK2 phosphorylation of CCA1 is necessary for its circadian oscillator function in Arabidopsis. Proc Natl Acad Sci USA 101: 3292-3297.
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355-365.
- Brognard J, Hunter T (2011) Protein kinase signaling networks in cancer. Curr Opin Genet Dev 21: 4-11.
- Zandomeni R, Zandomeni RC, Shugar D, Weinmann R (1986) Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem 261: 3414-3420.
- Sarno S, De Moliner E, Ruzzene M, Pagano MA, Battistutta R, et al. (2003) Biochemical and 3D-structural data on the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a) quinazolin-7-yl] acetic acid (IQA). Biochem J 374: 639-646.
- Sarno S, Salvi M, Battistutta R, Zanotti G, Pinna LA (2005) Features and potentials of ATP-site directed CK2 inhibitors. Biochim Biophys Acta 1754: 263-270.
- Agullo G, Gamet-Payrastre L, Manenti S, Viala C, Rémésy C, et al. (1997) Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition, Biochem Pharmacol 53: 1649-1657.
- Min L, Shuying M, Yueli T, Xiaoyun Z, Honglin Z, et al. (2015) Structural insights into avones as protein kinase CK2 inhibitors derived from a combined computational study. RSC Adv 5: 462.
- Souhila BT, Amel TM, Boubekeur M, Safia TK (2010) Modeling the binding modes of stilbene analogs to cyclooxygenase-2: a molecular docking study. J Mol Model 16: 1919-1929.
- Souhila BT, Redouane T, SaÃÂa TK (2013) Receptor and ligand-based 3D-QSAR study on a series of nonsteroidal anti-in¡ammatory drugs. Med Chem Res 22: 1529-1537.
- Rarey M, Kramer B, Lengauer T, Klebe G (1996) A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261: 470-489.
- Wolber G, Seidel T, Bendix F, Langer T (2008) Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today 13: 23-29.
- Yang SY (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 15: 444-450.
- Golub AG, Bdzhola VG, Ostrynska OV, Kyshenia IV, Sapelkin VM, et al. (20130) Discovery and characterization of synthetic 4’-hydroxyflavones-New CK2 inhibitors from flavone family. Bioorganic & Medicinal Chemistry 21: 6681-6689.
- MDL Information Systems (2002) ISIS/Draw 2.5. MDL Information System, San Leandro.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) USCF Chimera - A visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612.
- Lolli G, Cozza G, Mazzorana M, Tibaldi E, Cesaro L, et al. (2012) Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight Biochemistry 51: 6097-6107.
- Stierand K, Rarey M (2007) From modeling to medicinal chemistry: automatic generation of two-dimensional complex diagrams. Chem Med Chem 2: 853-860.
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, et al. (2006) A Critical Assessment of Docking Programs and Scoring Functions. J Med Chem 49: 5912-5931.
- Mukesh B, Rakesh K (2011) Molecular Docking: a Review. International Journal of Research in Ayurveda and Pharmacy 2: 1746-1751.
- Böhm HJ (1992) The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des 6: 61-78.
- Böhm HJ (1994) The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J Comput Aided Mol Des 8: 243-256.
- Rarey M, Kramer B, Lengauer T (1997) Multiple automatic base selection: protein-ligand docking based on incremental construction without manual intervention. J Comput Aided Mol Des 11: 369-384.
- Böhm HJ (1998) Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput-Aided Mol Des 12: 309-309.
- Wolber G, Langer T (2005) LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model 45: 160-169.
- Battistutta R, Mazzorana M, Cendron L, Bortolato A, Sarno S, et al. The ATP-Binding Site of Protein Kinase CK2 Holds a Positive Electrostatic Area and Conserved Water Molecules. Chem Bio Chem 8: 1804-1809.
- Sarno S, Papinutto E, Franchin C, Bain J, Elliott M, et al. (2011) ATP Site-Directed Inhibitors of Protein Kinase CK2: An Update. Curr Top Med Chem 11: 1340-1351.


