Keywords
Purα, Egr-1, amyloid precursor protein, transcriptional regulation, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD) is a type of irreversible degenerative disorder of the central nervous system that can induce a decline in intelligence and develop into progressing dementia. These effects are due to pathological damage, as illustrated by the deposition of the senile plaque, which is composed of 39-42 amino acids and denoted the amyloid β-peptide (Aβ), outside nervous cells [1-3]. Aβ originates from the amyloid precursor protein (APP) after a series of abnormal proteolytic degradations. To date, a transgenic mouse model for use in AD research has been successfully established [2].
APP is a type of transmembrane protein that is ubiquitously found inside the human body, and the gene encoding this protein is located on the 21th human chromosome. The accumulated evidence demonstrates that APP plays an important role in the development of Alzheimer’s disease (AD) because the amyloid β peptide originates from the cleavage of the amyloid precursor protein [4]. It has been proven that an increasing expression of APP accelerates the pathological development of AD [5,6]. Down’s Syndrome patients have an additional copy of the APP gene, which is also found in early onset AD-like pathology [7], and some AD patients also present an overexpression of APP in certain areas of the brain [8]. In contrast, APP is a widely expressed protein that is highly conserved throughout evolution and is developmentally regulated in parallel with synaptogenesis [9]. APP-knockout mice present deficiencies in postnatal growth, locomotor activity and grip strength, diminished hippocampal neuron viability, and retarded neuron development [10,11].
Actually, APP is a widely expressed protein that is highly conserved throughout evolution and is developmentally regulated in parallel with synaptogenesis [9]. The peaking point of APP gene expression levels in mice appeared at the second week of life and this just coincides with the summit of synaptogenesis then gradually decreases to low levels hereafter. Mattson MP et al reported that although there are multiple isoforms of APP, the expression pattern of the isoform may not be uniform throughout the mammalian nervous system [4]. APP promotes neurite outgrowth and synaptogenesis, modulates neuronal excitability and synaptic plasticity, all of these have been reported in in vitro researches and demonstrated that APP may play a protective effects on the neurons against oxidative stress [4]. APP is detected in various cells of most tissues and its expression is regulated by many cytokine-mediated factors, including growth factors [12], phorbol esters [13,14], and the super ligand family of nucleic receptors of the steroid/thyroid hormone [15,16]. The other reported transcription factors that regulate APP gene expression include Sp1 [17], p53 [18], Rac-1 [19], c/EBP [3] and p25/CDK5 [20].
Egr-1 is an early growth response protein and a member of the zinc finger family of transcription factors that displays Fos-like induction kinetics in many cells, including neurons. The Egr- 1 gene is located in 5q23-q31 of the human chromosome and encodes two exons. It has three zinc finger domains with a Cys2- His2 structure in their DNA-binding area, and its subunits always preferentially bind to GC-rich areas in a DNA sequence [21]. Egr-1 is an important regulator in the body because it controls the expression of many genes. Egr-1 regulates the transcription of late-response genes important for the synaptic plasticity processes, particularly the maintenance of long-term potentiation [22]. In addition, Egr-1 upregulates presenilin-2 gene expression in neuronal cells [23] and consequently the γ-secretase cleavage of APP. Moreover, Egr-1 is upregulated in the brain of patients with Alzheimer’s disease (AD), and the overexpression of Egr- 1 controls both the phosphorylation and dephosphorylation of tau by activating CDK5 and inactivating PP1, which leads to tau hyperphosphorylation and destabilized microtubules [24].
Purα is another highly conserved developmentally regulated protein which is ubiquitously existed almost in all metazoan animals. An increasing number of researches demonstrate that Purα plays a critical role in neuronal development and synaptogenesis. As a multifunctional protein, Purα binds to single stranded nucleic acids in a sequence specific manner. The role of Purα in cell survival and differentiation has been demonstrated in an animal model. Being a transcriptional factors, Purα can bind to the purine-rich sequences of DNA with a special way by which it can identify the specific sequences like (GGN)n in the DNA sequence [25]. The model of Purα knock out transgenic mice has been successfully established and it provides a perfect model for the research of the biological functions of Purα in many disciplines [26]. Our previous study successfully discovered that Pura acted as a negative regulator for APP gene expression, and primarily investigated the mechanism underlying the function of this protein [27]. An accumulated data have recognized that Purα plays important roles in DNA duplication and transcription and demonstrated that Purα is a crucial element in RNAcompartmentalized translation [28]. Our previous work has approved that Purα is a negative regulator for APP gene expression [27]. According to the examination of the APP promoter DNA sequence and computer-aided analysis, we found a (GGN) n region in the APP promoter 5’-untranslation region (5’-UTR) [27]. A series of experiments were designed to affirm that this site is the Purα-binding site with gel-shift and ChIP’s assays. Aided with reporter gene analysis, western blotting and histoimmunological analysis, we confirmed the negative regulatory effects of Purα on the APP gene expression.
APP promoters, both in human and mouse, lack traditional TATA and CCAAT box motifs. A number of researches have described the many GC rich sequences harbor in the APP promoter and a significant overlap between human and mouse APP promoters [29-31]. Several research groups have already exhibited a number of positive and negative regulatory elements within the human, mouse and rat APP promoter [29,30,32]. Considering the conserved nature of the GC rich sequences within both the human and mouse APP promoters, the inverse correlation in brain APP and Purα protein levels, and the known function of Purα and Egr-1 as transcriptional regulators, and their function in the nervous system, we sought to determine the mechanism of how Purα negatively regulates APP gene expression. In the subsequent studies, we have consistently found that Purα strongly down regulates APP gene expression and its interaction with Egr-1, would be helpful in elucidating the mechanism underlying the regulation of APP gene expression and its function in the development of AD. Therefore, the present study provides the first demonstration of the interaction between two transcriptional factors that have opposite regulative functions on APP gene expression, namely the negative regulator Purα and the positive regulator Egr-1. In this study, the APP promoter is regarded as the entry point, and APP gene expression and posttranscriptional regulation is assessed to analyze the possible action of APP expression in AD development. In addition, through investigation of the interaction between two different transcriptional factors, the regulative function of Purα on APP gene expression was illustrated, which may open a new avenue and provide insights for the prevention and treatment of AD.
Experimental Procedures
Chemicals and antibodies
Trichostatin A (TSA), butyrate, and suramin were purchased from Sigma-Aldrich Company (USA), and all of the chemicals were analytically pure. Rabbit anti-Egr-1 antibody was purchased from Cell Signaling Technology (USA), and HRP-conjugated goat antirabbit IgG was purchased from BIOSS Co. (Beijing, China). Mouse monoclonal antibody generated against the recombinant aa 66– 81 of APP, which recognizes the N-terminal part of the three major APP isoforms was purchased from Chemicon International (clone 22C11), and mouse monoclonal anti-Purα antibody, which was used to detect Purα, was purchased from Millipore (clone 2B7). Mouse anti-Grb2 antibody was purchased from BD Transduction laboratories. Dual-Luciferase® Reporter (DLR™) Assay System for luciferase assay was purchased from Promega Company (USA), transfection reagent LipofectamineTM 2000, Opt-MEM and DMEM medium were all purchased from InvitrogenTM (USA). LightShift® Chemiluminescent EMSA Kit for EMSA assay was purchased from Thermo (USA)
Plasmids and reporter constructs
The reporter plasmid constructs of the APP promoter in the pGL3 basic luciferase vector (Promega) utilized in this study are described in our previous published works [27], and the human APP promoter (bp−800/+118) was amplified by PCR from genomic DNA isolated from the human glioblastoma cell line (U87- MG) using primers representing bp 8201–8225 and 9188–9095 of the human APP promoter as listed below (refer to gi:2429080 for the complete sequence of the human APP gene). The mutant deletions of the APP promoter (-100 to +147), which lack the overlapping Purα (+63 to +77) and Egr-1 (+66 to +82) binding sites, were constructed by three-step PCR amplifications, and we used three pairs of primers to complete the amplification. First, we amplified the APP promoter fragment from -100 to +62 and +63 to +147 using the following PCR primers: forward primer p100, 5'-GGGGTACCGGCGGCGCCGCTAGGGTC-3’; reverse primer p62, 5’-GTCTCCCGGGGCCCCCGCGCAC-3’; forward primer p63, 5’-GGGCAGAGCAAAGGACGCGGCG-3’; and reverse primer p147, 5’-TTATAAATGTCGTTCGCGGGCGCA-3’. We then used the following primers to connect the two fragments in order to form the deleted mutant APP promoter ΔAPP100-147: forward, 5’-CGCCGCGTCCTTGCTCTGCCCGTCTCCCGGGGCCCCCGCGCAC- 3’, and reverse, TGCGCGGGGGCCCCGGGAGACGGGCAGAGCAAGGACGCGGCG- 3’. All of the PCR fragments were sub-cloned into the TOPO TA cloning vector (Invitrogen), digested with KpnI and XhoI, and sub-cloned into the KpnI and XhoI sites of the pGL3 basic vector. In the same way we amplified Egr-1 promoter fragment which is a 700 bp sequence spanned from -600 to +100 (4570 to 5270, refer to NCBI Reference Sequence: NG_021374.1) in the Egr-1 promoter with primers: forward: 5’-CGGGGTACCCATATAAGGAGCAGGAAGGA- 3’, revers, 5’-CCGCTCGAGCCTGGACGAGCAGGCTGGAG- 3’ from genomic DNA extracted from the human glioblastoma cell line (U87MG). The total of 700 bp fragment was inserted into pGL3 basic vector to construct the Egr-1 promoter reporter constructs pGL3-Egr-1(-600/+100). All the constructs were verified by sequencing. pCDNA3-Egr-1, pCDNA3-Purα, CMVp53 and mutant CMVp53 were maintained in our laboratory.
Cell culture, transient transfection of plasmids and siRNA, and luciferase assay
The human glioblastoma cell lines U87-MG, U251, and HEK293 and the human cervical carcinoma cell line HeLa were maintained in DMEM supplemented with 10% fetal bovine serum. Mouse embryo fibroblasts (MEFs) from mice presenting a targeted disruption of PURA or their wild-type littermates were prepared individually from embryos at gestation day 17 and maintained in DMEM supplemented with 10% fetal bovine serum. MEF cells were prepared and genotyped by PCR as previously described (25). The transient transfection of plasmid DNA into U87-MG and HeLa cells was performed with lipofectamineTM 2000 according to the manufacturer’s instructions. In short, cells were seeded in 24-well culture plates as 2 X 105 cells per well 24 hours before transfection. 2 hours before the transfection, to change the medium which is no antibiotics added. To dilute the lipofectamineTM 2000 and plasmid DNA with Opti-MEM (the ratio of DNA to lipofectamineTM 2000 (1:2 or 1:3) should be optimized before the experiment and we found 1:2.5 was the best-condition for our experiment). 1μg of plasmid DNA was diluted in 50μl of Opti-MEM and 2.5μl of lipofectamineTM 2000 was diluted in 50μl of Opti-MEM respectively and keep in room temperature for 5 minutes before mix the 2 dilutions to form the transfection mixture. The mixture was kept for 25 minutes before added to the cultured cells. We set the ratio of reporter plasmid to other eukaryotic expression plasmid, such as Purα and Egr- 1, as 1:1 and empty vector pCDNA3.0 was used to make up the total amount of DNA where necessary. 8 hour after the transfection, to change the medium and add the normal medium and keep the cells in cell incubator supplemented with 5% CO2 in 37°C. 48 hours after the transfection the cells were harvested and the cell lysates were prepared for the luciferase assay. MEF cells were transfected in a similar manner using 0.5 μg of the reporter constructs plus 0.5 μg of Pur-α expression construct and/or 0.5 μg of pCDNA3-Egr-1 plasmid per well in 24 well plate. For the HDAC assay, the cells were treated with 10 nM TSA or 1 nM butyrate 1 hour after transfection. The luciferase activity was normalized by the renilla activity. The results of triplicate (duplicate in the case of MEFs) samples in each experiment were averaged and presented as either fold increases over the control group or as relative light units (RLUs).
Separation of protein extracts and EMSA
Whole-cell extracts from U87-MG cells were prepared 36 hour after transfection with pCDNA3-Purα by lysing the cells in buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% NP-40 and protease inhibitors. An electrophoretic mobility shift assay (EMSA) was performed in a total reaction volume of 20 μl containing 20 μg of whole-cell extract and 60,000 cpm [γ−32P-ATP] end-labeled single stranded oligonucleotide probe in buffer containing 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 0.5 mM EDTA, 0.5 mM dithiothreitol, 1 mM MgCl2, and 4% glycerol. After incubation for 30 min on ice, the reactions were loaded on 8% native polyacrylamide gels and electrophoresed at 180V (20 mA) in 0.5x TBE buffer for 4 h. The gels were dried and visualized by autoradiography. For the competition and supershift assays, unlabeled competitor oligonucleotides, antibody, or normal serum were preincubated with the extracts overnight at 4°C in reaction buffer before addition of the probe. The APP probe used in this experiment represents the GC-rich sequence present at +63 to +89 of the human proximal APP promoter, namely 5’-acg gcg gtg gcg cgg gca ga-3’, whereas the non-specific competitor oligonucleotide sequence comprised the following A/T rich sequence: 5’-tct gta cgt gac cac act cac ctc-3’. For alternate EMSA assay, the oligo was labeled with digoxin and performed according to the manufacturer’s instruction.
Western blotting
The U87MG cells, HeLa cells, transfected cells and TSA- and butyrate-treated cells (U87MG and HeLa cells treated with 10 nM TSA, 1 mM butyrate and 300 nM suramin for 4 and 6 h, respectively) were washed twice with ice-cold PBS and trypsinized, and the cell pellets were lysed in RIPA buffer (1x PBS plus 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, and protease inhibitors). The lysates were sonicated and centrifuged to remove any insoluble debris. The protein concentration of the supernatants was normalized using the Bradford assay (Bio-Rad), and 50 μg of the extracts was analyzed by 10% SDS-PAGE. The proteins were transferred to nitrocellulose membranes, and the membranes were blocked with 5% nonfat dry milk in 1x PBST for 1 h and incubated with primary antibody overnight at 4°C. The membranes were then washed in 1x PBST and incubated in the appropriate horseradish peroxidase-conjugated anti-mouse secondary antibodies for 1 h, and the proteins were detected by ECL-Plus according to the manufacturer’s instructions (Amersham). To detect APP, a mouse monoclonal antibody generated against the recombinant aa 66–81 of APP (dilution 1:1,000), which recognizes the N-terminal part of the three major APP isoforms (clone 22C11, Chemicon International), was used. A mouse monoclonal anti-Purα antibody, which was described previously, was used to detect Purα (clone 10B12, see [25] for details). Mouse anti-Myc tag (Invitrogen, dilution 1:1,000) was used to detect Purα in the cells transfected with the Myc-tagged Purα construct. To detect Egr- 1, a rabbit anti-human Egr-1 antibody (Cell Signaling Technology, 1:1000) was used. A mouse anti-Grb2 antibody (BD Transduction laboratories, dilution 1:1,000) was also used to verify the loading conditions for western blotting.
Chromatin immune precipitation (ChIP) assay
The cells were transfected with the APP promoter constructs –143 to +118, and 48 h after transfection, the cells were crosslinked with formaldehyde, which was added to the culture media to a final concentration of 1%, and incubated for 10 min. The cells were then washed briefly in ice-cold PBS containing protease inhibitors, and the cell lysates were scraped and then sonicated to shear the DNA. The cell supernatants were collected and precleared with salmon sperm DNA/protein-A agarose for 30 min with rotation. Immunoprecipitation was then performed using a polyclonal antibody to Purα or normal rabbit serum overnight at 4°C with rotation. The agarose beads were then pelleted, the pellets were washed, and the cross-linked protein-DNA complexes were eluted by reversing the histone-DNA crosslinks through heating for 2 h at 65°C in 200 mM NaCl. PCR was then performed using the following primer pairs: forward 5’-ggg gcg cga ggg ccc ctc cc-3’, reverse 5’-tgc tgt gcg agt ggg atc cgc gtc ctt- 3’. The PCR product should be 260 bp and spanned from (−143 to +118) 8859 to 9118.
Immunocytochemistry
The cells were placed on poly-L-lysine-coated glass chamber slides and allowed to attach overnight. U87MG cells were transfected with 1.0 μg of Purα and Egr-1 expression plasmids. The cells were then fixed for 3 min in ice-cold acetone and washed with PBS. After blocking with 2% normal rabbit serum for 2 h, the slides were double-labeled by incubation with primary antibodies (Purα and Egr-1) overnight at room temperature. The cells were then washed with PBS, incubated with anti-rabbit Egr- 1 or FITC-conjugated Purα secondary antibodies for 2 h at room temperature in the dark, rinsed with PBS, and mounted in an aqueous mounting medium (Vector Laboratories, Burlingame, CA, USA).
Statistical analysis
Data were statistically analyzed with unpaired Student’s t-test with Welch correction depending on population (GraphPad In Start 3.0; GraphPad Software, San Diego, CA, USA) and presented as mean ± SEM. Significance levels were labeled as *p<0.05, **p<0.01 and ***p<0.001.
Results
The binding sites of Egr-1 and Purα in the 5’-UTR region of the APP promoter determine the role of these two transcriptional factors in the regulation of APP gene expression
By carefully reviewing the DNA sequence in the proximal end and 5’-UTR of the APP promoter, we noted that the existence of Egr-1 and Purα binding sites in the 5’-UTR of the APP promoter and they are overlapped. We found that the sequence with characteristics of a Purα-binding site was located in +63 to +77 and that there are five repeats of the GGN structure existed. In addition, the Egr- 1-binding site is close to this site and overlaps the Purα binding site: these two sites are located in +63 to +79 and +66 to +82 of the APP promoter 5’-UTR ( Figure 1).
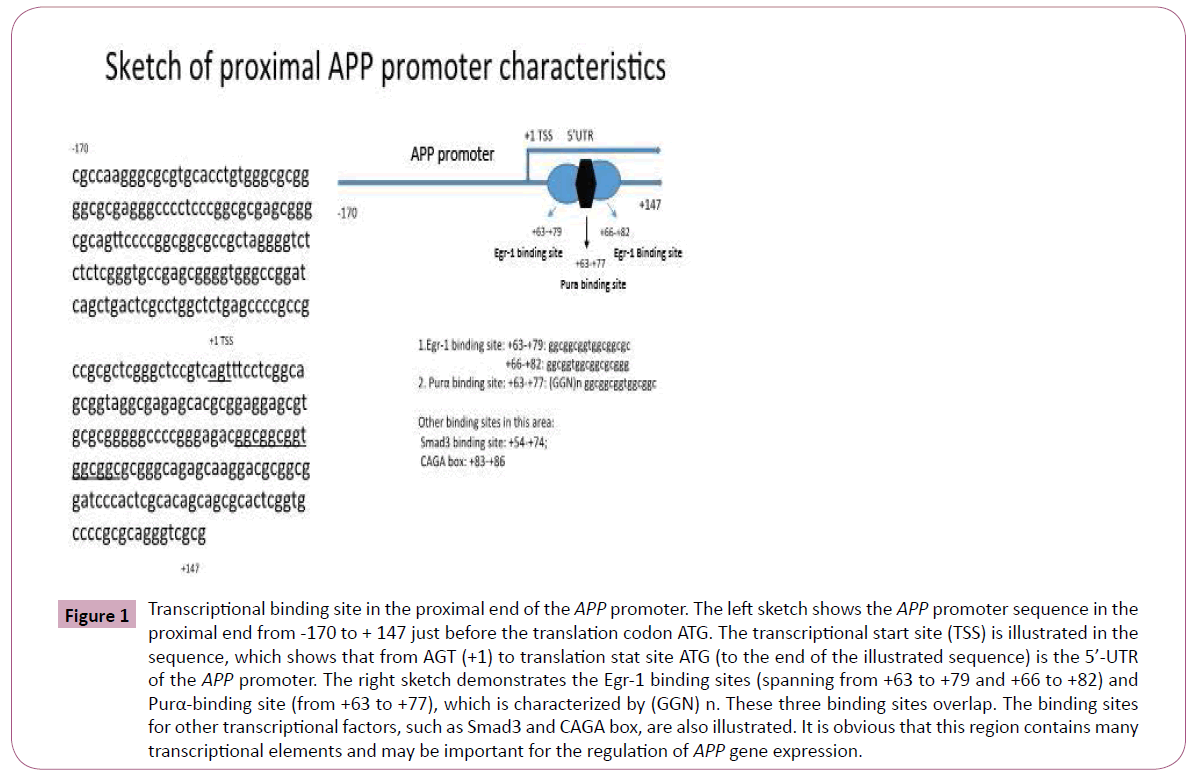
Figure 1: Transcriptional binding site in the proximal end of the APP promoter. The left sketch shows the APP promoter sequence in the proximal end from -170 to + 147 just before the translation codon ATG. The transcriptional start site (TSS) is illustrated in the sequence, which shows that from AGT (+1) to translation stat site ATG (to the end of the illustrated sequence) is the 5’-UTR of the APP promoter. The right sketch demonstrates the Egr-1 binding sites (spanning from +63 to +79 and +66 to +82) and Purα-binding site (from +63 to +77), which is characterized by (GGN) n. These three binding sites overlap. The binding sites for other transcriptional factors, such as Smad3 and CAGA box, are also illustrated. It is obvious that this region contains many transcriptional elements and may be important for the regulation of APP gene expression.
To evaluate the effects of Egr-1 and Purα on the regulation of APP promoter activities, we co-transfected U87MG cells with the APP promoter reporter constructs pGL3-APP-91/+118 and pGL3-APP-91/+1 with the Purα or Egr-1 eukaryotic expression construct, and 48 h after transfection, the cell extracts were collected and subjected to a luciferase assay to evaluate the effects of these two transcriptional factors on the APP promoter activities. The results obtained from the co-transfection of APP- 91/+118 and APP-91/+1 into U87MG cells demonstrated that Purα can downregulate APP promoter activities and that Egr- 1 can upregulate APP promoter activities compared with the control group (the APP plus pCDNA3 vector instead of Purα and Egr-1), and these differences are statistically significant (p < 0.01, n = 6, Figure 2).
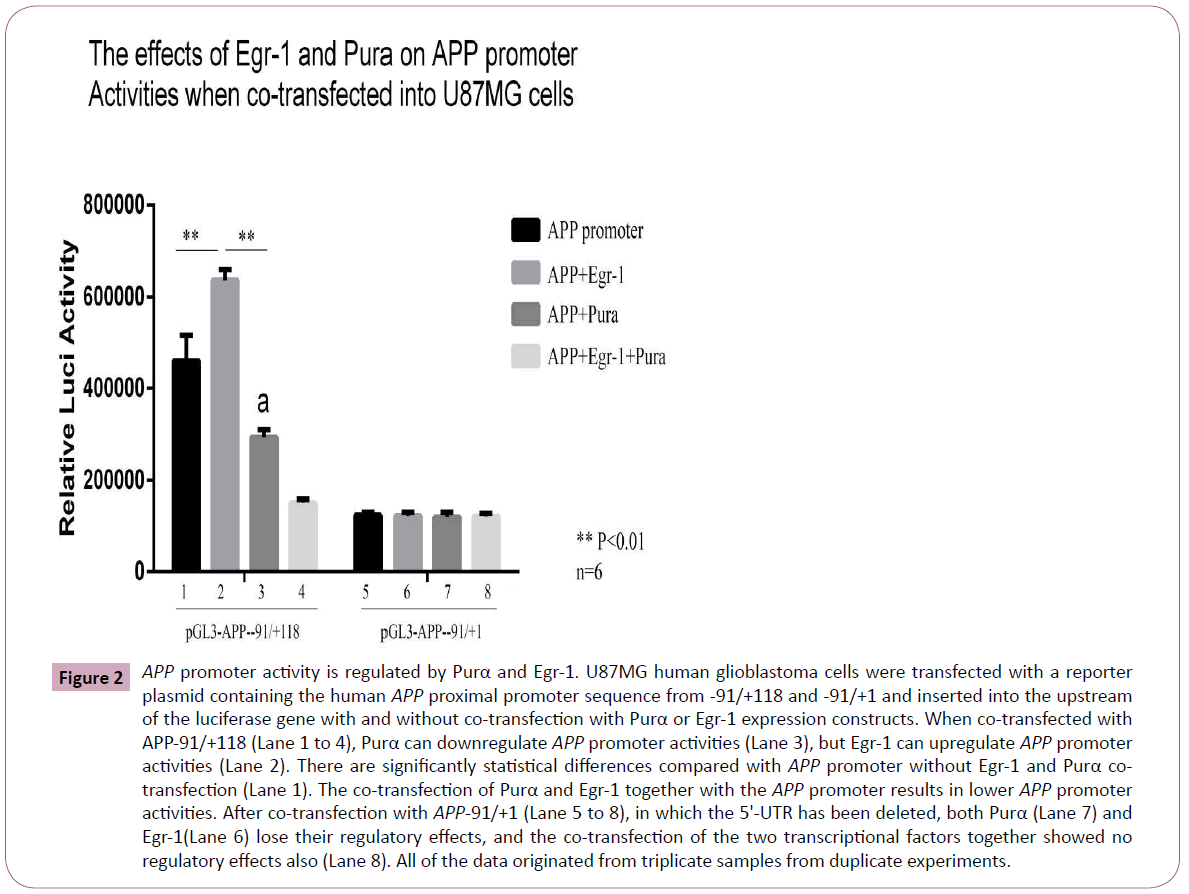
Figure 2: APP promoter activity is regulated by Purα and Egr-1. U87MG human glioblastoma cells were transfected with a reporter plasmid containing the human APP proximal promoter sequence from -91/+118 and -91/+1 and inserted into the upstream of the luciferase gene with and without co-transfection with Purα or Egr-1 expression constructs. When co-transfected with APP-91/+118 (Lane 1 to 4), Purα can downregulate APP promoter activities (Lane 3), but Egr-1 can upregulate APP promoter activities (Lane 2). There are significantly statistical differences compared with APP promoter without Egr-1 and Purα cotransfection (Lane 1). The co-transfection of Purα and Egr-1 together with the APP promoter results in lower APP promoter activities. After co-transfection with APP-91/+1 (Lane 5 to 8), in which the 5'-UTR has been deleted, both Purα (Lane 7) and Egr-1(Lane 6) lose their regulatory effects, and the co-transfection of the two transcriptional factors together showed no regulatory effects also (Lane 8). All of the data originated from triplicate samples from duplicate experiments.
To further assess the effects of Purα and Egr-1 on APP promoter activities, and Purα-knockout mouse embryo fibroblast cells (MEFs) were used for the transfection to check the effects of Purα on APP promoter activities. We co-transfected the Egr-1 eukaryotic expression vector, pCDNA3-Egr1, together with luciferase reporter constructs of APP promoter pGL3-APP-91/+118 and 5’- UTR deleted mutation pGL3-APP-91/+1 into the mouse embryo fibroblast cell lines (MEFs) originated from the Purα knock out mice, Purα-/- and their wild-type Purα+/+ MEFs to assess the effects of endogenous Purα on Egr-1 as well as APP promoter activities. The luciferase assay results demonstrated that the transfection of -91/+118 into Purα-/- cells markedly increased the effects of Egr-1 on APP promoter activities compared with its transfection into Purα+/+ cells, the promoter activities increased by1.8-fold, whereas in Purα+/+ cells, the activities increased only by 40.6%. This finding indicates that the endogenous Purα reduced the effects of Egr-1 on the APP promoter: in cells in which Purα has been knocked out, the effects of Purα is not existed, the effects of Purα on Egr-1 has been relieved, in this circumstances Egr-1 can exhibit maximum upregulation effects on APP promoter activities. In contrast, after the deletion of the 5’-UTR, Egr-1 has no up-regulatory effects on the APP promoter at all no matter in Purα-/- MEFs or Purα+/+ MEFs because there are no Egr-1 binding sites in this region of the AβPP promoter (Figure 3).
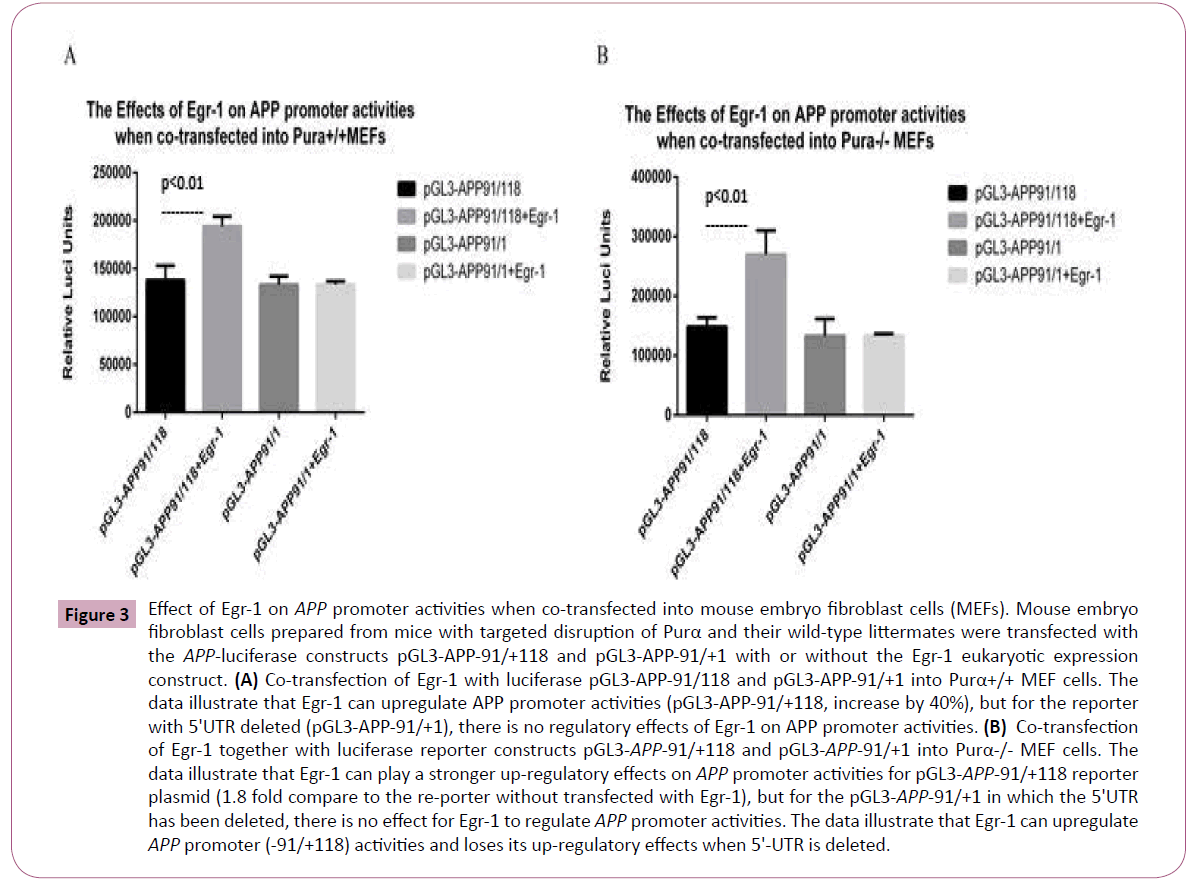
Figure 3: Effect of Egr-1 on APP promoter activities when co-transfected into mouse embryo fibroblast cells (MEFs). Mouse embryo fibroblast cells prepared from mice with targeted disruption of Purα and their wild-type littermates were transfected with the APP-luciferase constructs pGL3-APP-91/+118 and pGL3-APP-91/+1 with or without the Egr-1 eukaryotic expression construct. (A) Co-transfection of Egr-1 with luciferase pGL3-APP-91/118 and pGL3-APP-91/+1 into Purα+/+ MEF cells. The data illustrate that Egr-1 can upregulate APP promoter activities (pGL3-APP-91/+118, increase by 40%), but for the reporter with 5'UTR deleted (pGL3-APP-91/+1), there is no regulatory effects of Egr-1 on APP promoter activities. (B) Co-transfection of Egr-1 together with luciferase reporter constructs pGL3-APP-91/+118 and pGL3-APP-91/+1 into Purα-/- MEF cells. The data illustrate that Egr-1 can play a stronger up-regulatory effects on APP promoter activities for pGL3-APP-91/+118 reporter plasmid (1.8 fold compare to the re-porter without transfected with Egr-1), but for the pGL3-APP-91/+1 in which the 5'UTR has been deleted, there is no effect for Egr-1 to regulate APP promoter activities. The data illustrate that Egr-1 can upregulate APP promoter (-91/+118) activities and loses its up-regulatory effects when 5'-UTR is deleted.
Mapping the Purα and Egr-1 binding sites within the APP promoter
Based on an analysis of the APP promoter and the promoter deletion mapping studies performed in this study, we identified several potential binding sites for both Egr-1 and Pur-α within critical regions, and these binding sites appeared to be overlapped. To simplify the complexity of the APP promoter and exclude other influencing factors, simply deleting the 5’-UTR is not sufficient because there are other binding sites for TFs in this region. Therefore, we deleted the overlapping Egr-1- and Purα- binding sites, which spanned from +63 to +82, and maintained the other sequences of the 5’UTR to construct the following APP reporter genes: pGL3-APP-100/+147 mutant and the wild-type pGL3-APP-100/+147. The two constructs were co-transfected separately with Egr-1 or Purα eukaryotic vector into 293HEK and U251MG cells to evaluate the effects of these two TFs on the APP promoter. The results demonstrated that Purα can still downregulate and Egr-1 can upregulate APP promoter activities in the 293HEK and U251MG cells; however, in the cells that had been transfected with the pGL3-APP-100/+147 mutant, which deletes these binding sites, both TFs lost their regulatory effects on APP promoter activity (Figure 4) because the only binding sites for Purα and Egr-1 in the region of the APP promoter from -100 to +147 are located in +63 to +82. In the cells in which this region has been deleted, the Purα- and Egr-1-binding sites in the APP promoter are deleted, and the regulatory functions of these two TFs are thus lost.
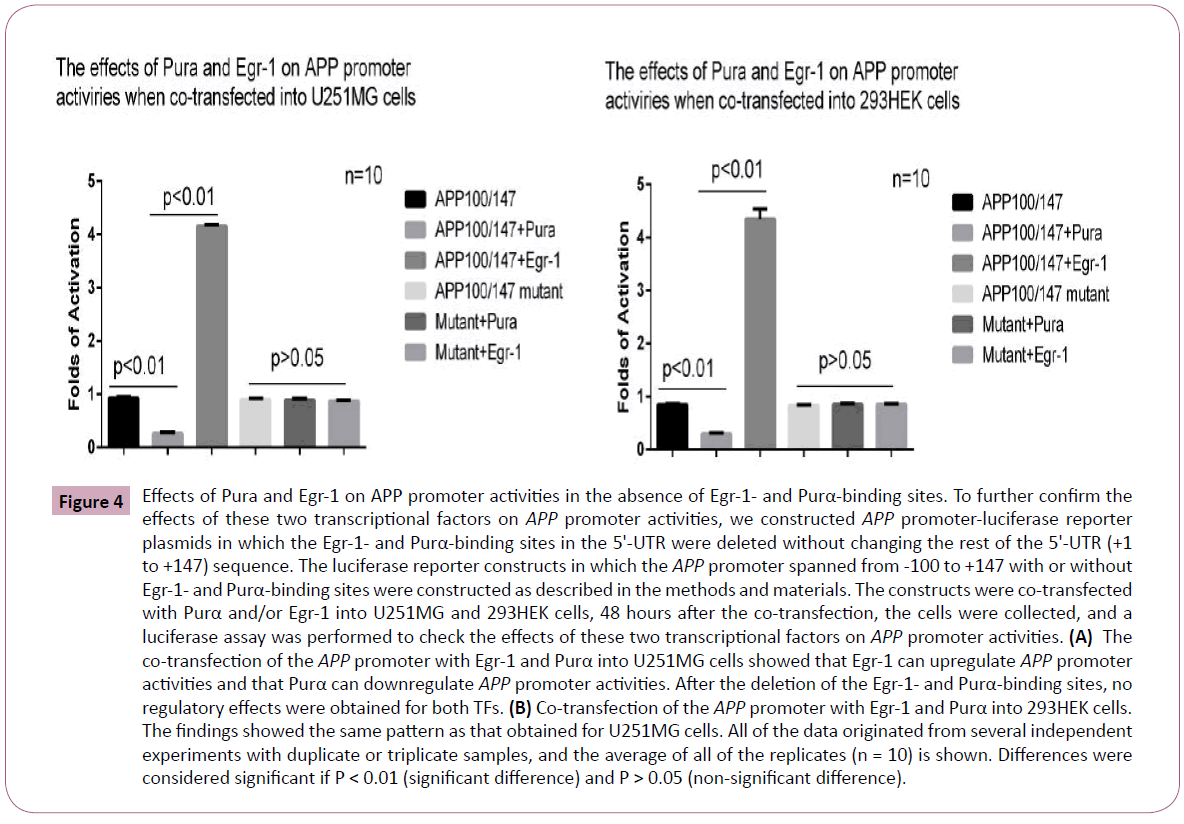
Figure 4: Effects of Pura and Egr-1 on APP promoter activities in the absence of Egr-1- and Purα-binding sites. To further confirm the effects of these two transcriptional factors on APP promoter activities, we constructed APP promoter-luciferase reporter plasmids in which the Egr-1- and Purα-binding sites in the 5'-UTR were deleted without changing the rest of the 5'-UTR (+1 to +147) sequence. The luciferase reporter constructs in which the APP promoter spanned from -100 to +147 with or without Egr-1- and Purα-binding sites were constructed as described in the methods and materials. The constructs were co-transfected with Purα and/or Egr-1 into U251MG and 293HEK cells, 48 hours after the co-transfection, the cells were collected, and a luciferase assay was performed to check the effects of these two transcriptional factors on APP promoter activities. (A) The co-transfection of the APP promoter with Egr-1 and Purα into U251MG cells showed that Egr-1 can upregulate APP promoter activities and that Purα can downregulate APP promoter activities. After the deletion of the Egr-1- and Purα-binding sites, no regulatory effects were obtained for both TFs. (B) Co-transfection of the APP promoter with Egr-1 and Purα into 293HEK cells. The findings showed the same pattern as that obtained for U251MG cells. All of the data originated from several independent experiments with duplicate or triplicate samples, and the average of all of the replicates (n = 10) is shown. Differences were considered significant if P < 0.01 (significant difference) and P > 0.05 (non-significant difference).
Confirmation of the binding of Egr-1 to the APP promoter by EMSA and ChIP assay
To further confirm the binding of these two TFs, we tested this interaction through ChIP’s assay by immunoprecipitation of cross-linked nuclear protein/DNA complexes with antibodies to Purα and Egr-1 followed by PCR using primer sets spanning the APP promoter. As shown in Figure 5A, we transfected glial cells with the Purα and Egr-1 eukaryotic expression constructs and then precipitated the two proteins in complex with the Purα and Egr-1 antibodies. APP promoter primers spanning the region from -143 to +118 were used for PCR to analyze the same DNA fragment in glial cells. The results demonstrated that the same DNA fragment could be amplified from the precipitated complex. However, when the two transcriptional factors were transfected together, the amplified bands appeared to be weaker than those obtained when only Purα or Egr-1 was transfected. This finding may indicate that the two transcriptional factors competed to bind to the same sites in the APP promoter sequence.
To further evaluate the promoter binding ability of these proteins, we analyzed the nuclear extracts from cells transfected with the Egr-1 and Purα expression constructs through EMSA using oligonucleotides representing the GC-rich region of the human proximal APP promoter, which overlaps the 5’-UTR (+63 to +87) (5'-cgcggcggtggcggcgcgggcaga-3’) and contains putative binding sites for both of these proteins. The results demonstrated that the Egr-1 nucleic extracts can bind to this oligo, and the intensity of the binding band increased with an increase in the amount of Egr-1 nucleic extracts. A close dose-intensity relationship was found between the NE amount and the intensity of the binding band. The addition of Egr-1 antibody and 100X cold concentrated oligo decreased the intensity of the binding band, but the nonspecific oligo did not compete with the hot oligo, which implies that the binding of Egr-1 to the oligo is specific. In addition, we also found that the nucleic extracts from Purα-transfected cells can also bind to the oligo, but the size of the binding band was smaller than that obtained with Egr-1 because the molecular weight of Purα is lower than that of Egr-1 (Figure 5B).
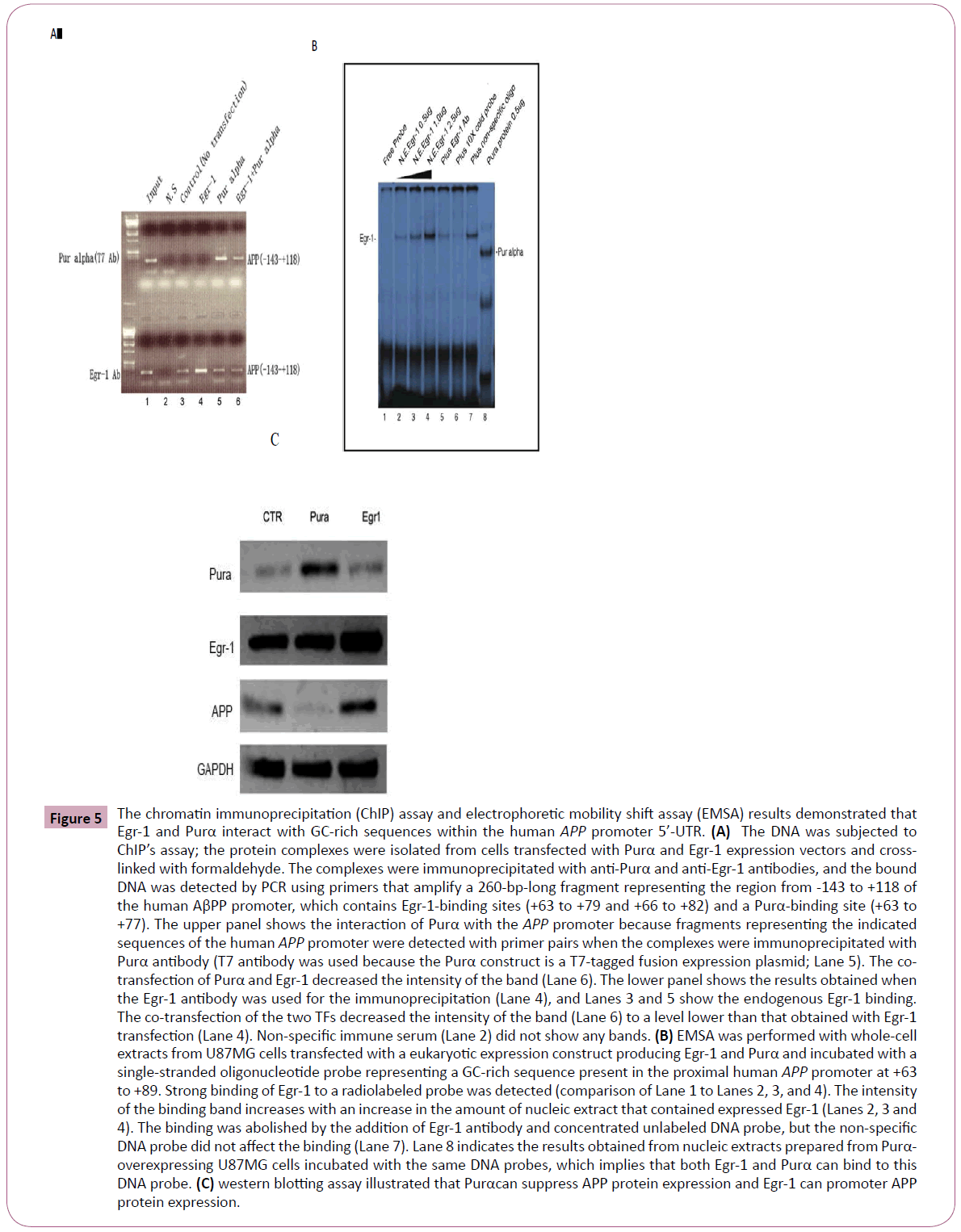
Figure 5: The chromatin immunoprecipitation (ChIP) assay and electrophoretic mobility shift assay (EMSA) results demonstrated that Egr-1 and Purα interact with GC-rich sequences within the human APP promoter 5’-UTR. (A) The DNA was subjected to ChIP’s assay; the protein complexes were isolated from cells transfected with Purα and Egr-1 expression vectors and crosslinked with formaldehyde. The complexes were immunoprecipitated with anti-Purα and anti-Egr-1 antibodies, and the bound DNA was detected by PCR using primers that amplify a 260-bp-long fragment representing the region from -143 to +118 of the human AβPP promoter, which contains Egr-1-binding sites (+63 to +79 and +66 to +82) and a Purα-binding site (+63 to +77). The upper panel shows the interaction of Purα with the APP promoter because fragments representing the indicated sequences of the human APP promoter were detected with primer pairs when the complexes were immunoprecipitated with Purα antibody (T7 antibody was used because the Purα construct is a T7-tagged fusion expression plasmid; Lane 5). The cotransfection of Purα and Egr-1 decreased the intensity of the band (Lane 6). The lower panel shows the results obtained when the Egr-1 antibody was used for the immunoprecipitation (Lane 4), and Lanes 3 and 5 show the endogenous Egr-1 binding. The co-transfection of the two TFs decreased the intensity of the band (Lane 6) to a level lower than that obtained with Egr-1 transfection (Lane 4). Non-specific immune serum (Lane 2) did not show any bands. (B) EMSA was performed with whole-cell extracts from U87MG cells transfected with a eukaryotic expression construct producing Egr-1 and Purα and incubated with a single-stranded oligonucleotide probe representing a GC-rich sequence present in the proximal human APP promoter at +63 to +89. Strong binding of Egr-1 to a radiolabeled probe was detected (comparison of Lane 1 to Lanes 2, 3, and 4). The intensity of the binding band increases with an increase in the amount of nucleic extract that contained expressed Egr-1 (Lanes 2, 3 and 4). The binding was abolished by the addition of Egr-1 antibody and concentrated unlabeled DNA probe, but the non-specific DNA probe did not affect the binding (Lane 7). Lane 8 indicates the results obtained from nucleic extracts prepared from Purα- overexpressing U87MG cells incubated with the same DNA probes, which implies that both Egr-1 and Purα can bind to this DNA probe. (C) western blotting assay illustrated that Purαcan suppress APP protein expression and Egr-1 can promoter APP protein expression.
HDAC inhibitor stimulates Egr-1 transcriptional activity
Histone deacetylase (HDAC) inhibitors have been utilized as epigenetic modifiers for the treatment of a number of CNS disorders, including epilepsy, schizophrenia, and Alzheimer's disease. The cells treated with HDAC inhibitor exhibited increased APP promoter transcriptional activities. We checked the effects of the HDAC inhibitors TSA and butyrate on APP promoter activities. One hour after U87MG and HeLa cells were transfected with the APP promoter, the cells were treated with 10 nM TSA or 1 nM butyrate, and 48 h after transfection, a luciferase assay was performed to evaluate the APP promoter activities. The results demonstrated that both TSA and butyrate can increase the transactivities of the APP promoter, but the efficiency was different between the two different cell lines (Figure 6A). HeLa cells demonstrated markedly increased transactivities compared with U87 MG cells, potentially due to differences in transfection efficiency (HeLa cells presented greater transfection efficiency than U87MG cells). Another reason for this difference may be due to the origin of the two cell lines: cells from a non-nervous origin are more sensitive to stimulation with HDAC inhibitor than cells with a nervous origin. It is striking that Purα can completely suppress the effects of the HDAC inhibitor induced upregulation on APP promoter transactivities because the co-transfection of the APP promoter with Purα under the same conditions and subsequent treatment with the HDAC inhibitor suppressed the increased transactivities of HDAC inhibitors in both U87MG and HeLa cells (Figure 6B).
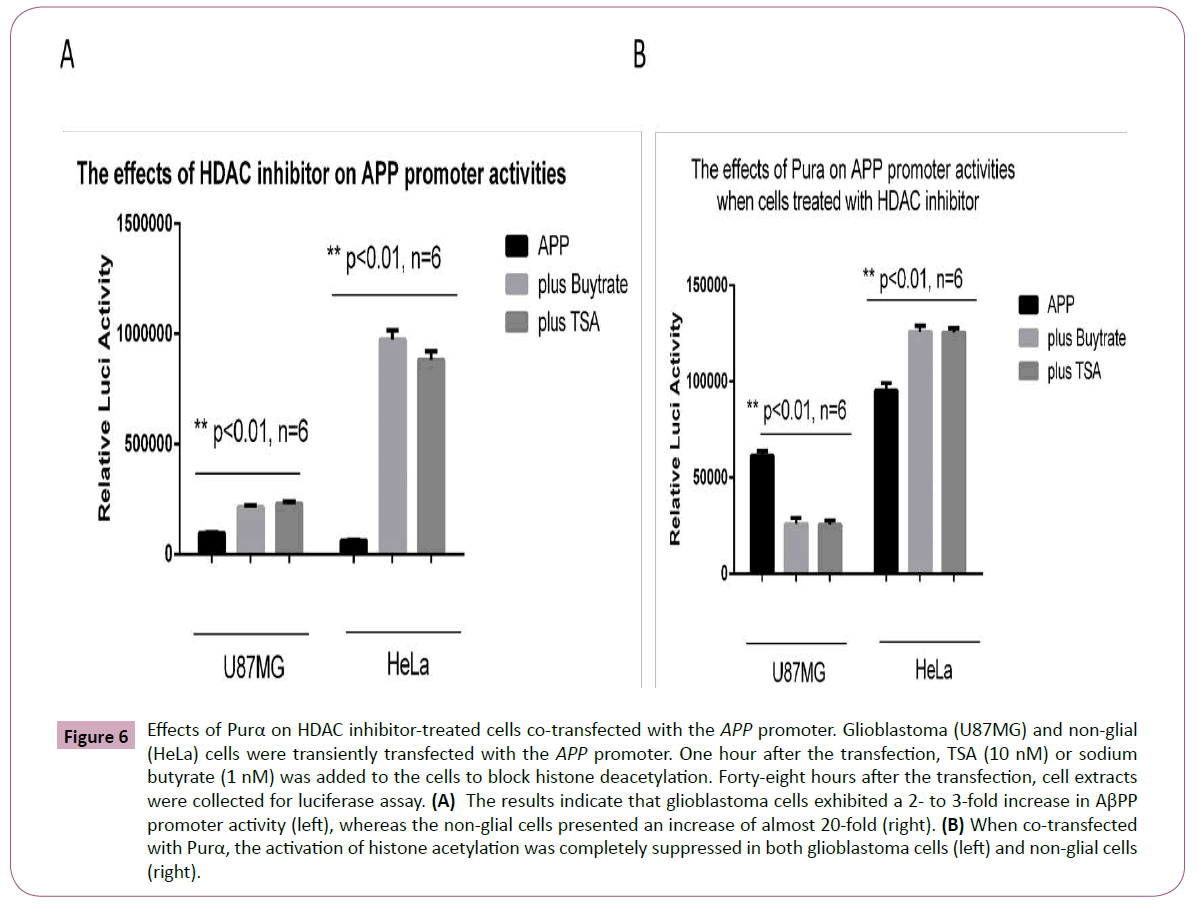
Figure 6: Effects of Purα on HDAC inhibitor-treated cells co-transfected with the APP promoter. Glioblastoma (U87MG) and non-glial (HeLa) cells were transiently transfected with the APP promoter. One hour after the transfection, TSA (10 nM) or sodium butyrate (1 nM) was added to the cells to block histone deacetylation. Forty-eight hours after the transfection, cell extracts were collected for luciferase assay. (A) The results indicate that glioblastoma cells exhibited a 2- to 3-fold increase in AβPP promoter activity (left), whereas the non-glial cells presented an increase of almost 20-fold (right). (B) When co-transfected with Purα, the activation of histone acetylation was completely suppressed in both glioblastoma cells (left) and non-glial cells (right).
To determine why Purα can suppress the effects of HDAC inhibitors on APP promoter activities, we investigated what happened inside the cells after treatment with the HDAC inhibitor. Western blot analysis was employed to evaluate the changes inside cells after HDAC inhibition. The results illustrated that treatment with HADC inhibitor markedly increased the endogenous level of Egr-1 expression (Figure 7A), which suggests that the HDAC inhibitor induces endogenous Egr-1 expression and thereby increase the APP promoter activities. The EMSA results confirmed that Egr-1 level was elevated after treatment with TSA. The cell nuclei extracts from U87MG and HeLa cells treated with TSA were collected, and EMSA was performed as previously described to show that the endogenous Egr-1 level increased (Figure 7B).
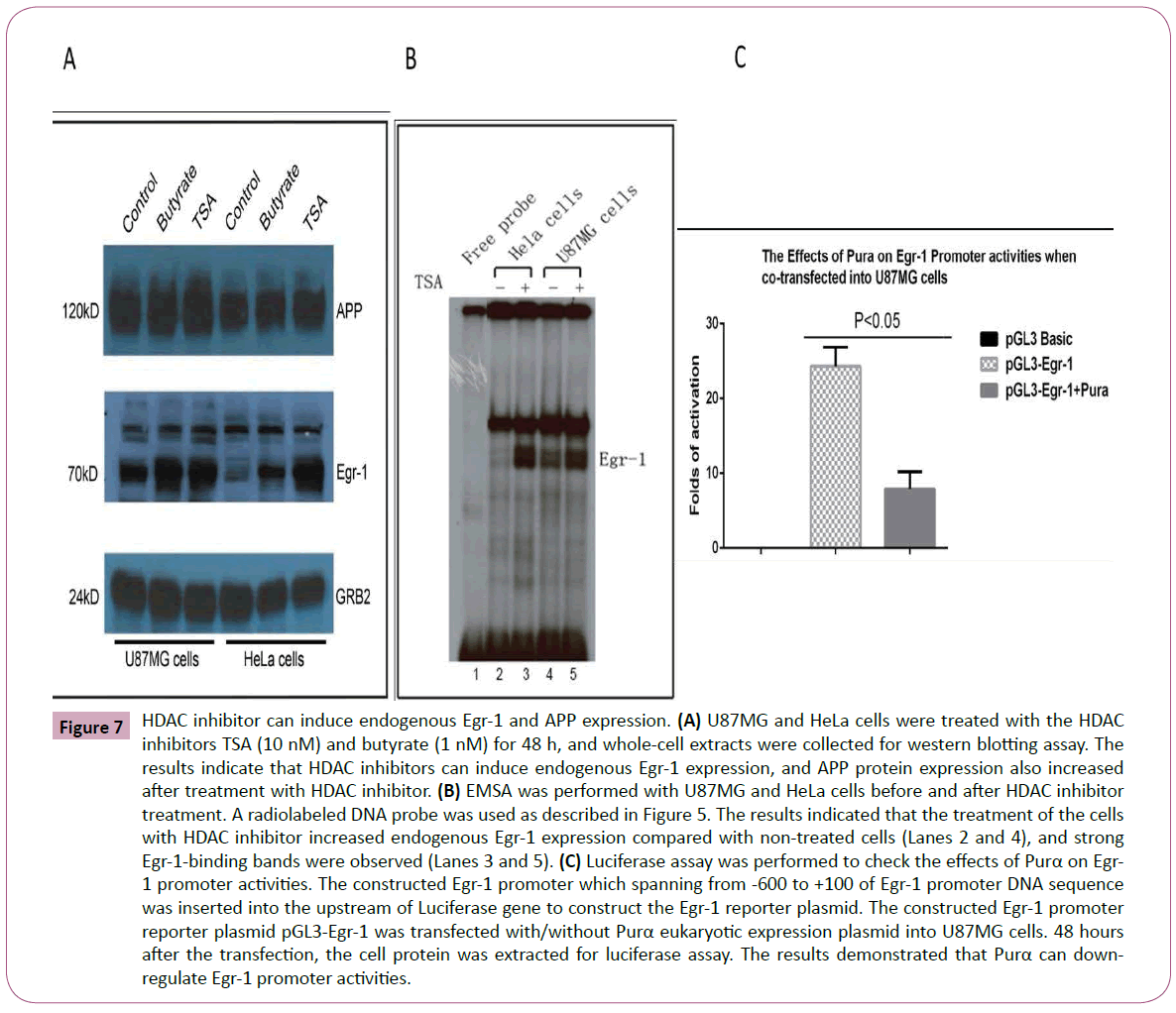
Figure 7: HDAC inhibitor can induce endogenous Egr-1 and APP expression. (A) U87MG and HeLa cells were treated with the HDAC inhibitors TSA (10 nM) and butyrate (1 nM) for 48 h, and whole-cell extracts were collected for western blotting assay. The results indicate that HDAC inhibitors can induce endogenous Egr-1 expression, and APP protein expression also increased after treatment with HDAC inhibitor. (B) EMSA was performed with U87MG and HeLa cells before and after HDAC inhibitor treatment. A radiolabeled DNA probe was used as described in Figure 5. The results indicated that the treatment of the cells with HDAC inhibitor increased endogenous Egr-1 expression compared with non-treated cells (Lanes 2 and 4), and strong Egr-1-binding bands were observed (Lanes 3 and 5). (C) Luciferase assay was performed to check the effects of Purα on Egr- 1 promoter activities. The constructed Egr-1 promoter which spanning from -600 to +100 of Egr-1 promoter DNA sequence was inserted into the upstream of Luciferase gene to construct the Egr-1 reporter plasmid. The constructed Egr-1 promoter reporter plasmid pGL3-Egr-1 was transfected with/without Purα eukaryotic expression plasmid into U87MG cells. 48 hours after the transfection, the cell protein was extracted for luciferase assay. The results demonstrated that Purα can downregulate Egr-1 promoter activities.
Purα can suppress endogenous Egr-1 expression
As described above, Purα and Egr-1 may interact in vivo, and it appears that Purα can suppress the function of Egr-1. Thus, it is necessary to determine whether Purα affects endogenous Egr- 1 expression. We used the HDAC inhibitors TSA and butyrate to induce endogenous Egr-1 expression, and the Egr-1 inhibitor suramin was used to analyze Egr-1 expression by western blotting. The results demonstrated that the HDAC inhibitor can increase Egr-1 expression in both U87MG and HeLa cells and that suramin can suppress Egr-1 expression (Figure 8A and Figure 8B). Significant differences were obtained between the cells treated with the HDAC inhibitors TSA and suramin (Figure 8E). In addition, a luciferase assay was performed to check the effects of suramin on APP promoter activities, and the results confirmed that the treatment of cells with suramin decreased the APP promoter activities (Figure 8D), which implies that suramin can successfully inhibit the effects of Egr-1 on APP promoter activities. To further analyze the effects of Purα on endogenous Egr-1 expression, we checked the effects of Purα on Egr-1 promoter activities through a luciferase assay. The constructed luciferase reporter plasmid with Egr-1 promoter inserted in the upstream of luciferase gene, pGL3-Basic-Egr-1 reporter which spanned from -600 to +100 in the Egr-1 promoter was used to co-transfect with Purα eukaryotic expression vector, pCDNA3-Purα into U87MG cells and the results demonstrated that Purα plays a negative regulatory effects on Egr-1 promoter transactivities. That implies that Purα can suppress the endogenous Egr-1 expression (Figure 7C).
To further confirm the findings we observed in the reporter assay, we employed another experiment to verify the effects of Purα on APP gene expression. We transfected the Purα eukaryotic expression constructs pCDNA3-Purα, wild-type p53, which is another Egr-1 activator, and a p53 mutant into U87MG cells and prepared cells extracts for western bolting. The results demonstrated that Purα can suppress endogenous Egr-1 expression, that p53 can increase the expression of endogenous Egr-1 in vivo and that the mutant p53 did not affect endogenous Egr-1 expression (Figure 8C) compared with the control group, and a significant difference was obtained between the Purα and p53 groups (Figure 8F). All of the above-mentioned results confirmed that Egr-1 can be a positive regulator of APP gene expression, that HDAC inhibitors increase Egr-1 and increase APP binding activity, and that these effects do not result in an increase in APP expression in the presence of Purα because Purα strongly suppresses APP promoter activity even in the presence of Egr-1.
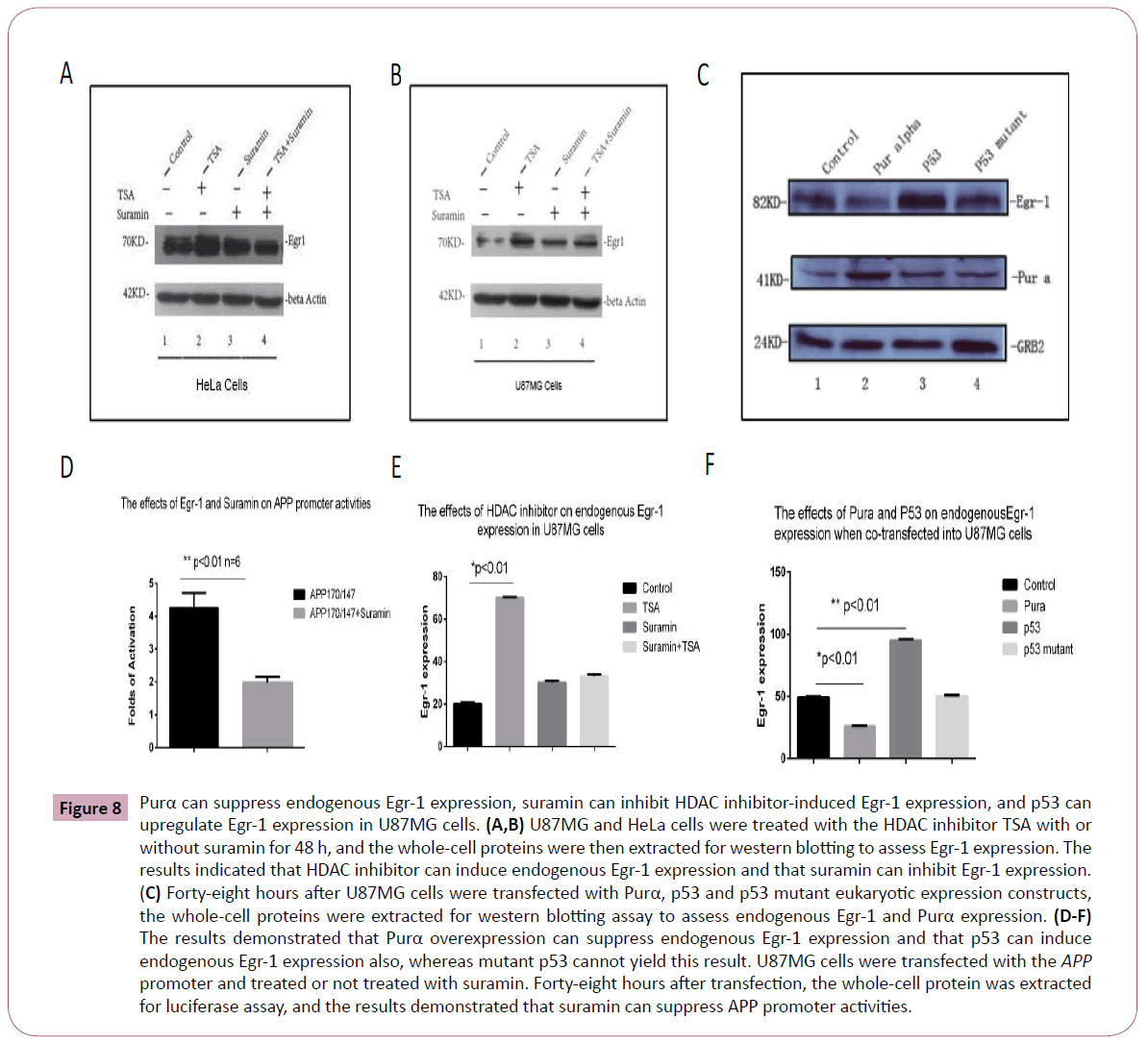
Figure 8: Purα can suppress endogenous Egr-1 expression, suramin can inhibit HDAC inhibitor-induced Egr-1 expression, and p53 can upregulate Egr-1 expression in U87MG cells. (A,B) U87MG and HeLa cells were treated with the HDAC inhibitor TSA with or without suramin for 48 h, and the whole-cell proteins were then extracted for western blotting to assess Egr-1 expression. The results indicated that HDAC inhibitor can induce endogenous Egr-1 expression and that suramin can inhibit Egr-1 expression. (C) Forty-eight hours after U87MG cells were transfected with Purα, p53 and p53 mutant eukaryotic expression constructs, the whole-cell proteins were extracted for western blotting assay to assess endogenous Egr-1 and Purα expression. (D-F) The results demonstrated that Purα overexpression can suppress endogenous Egr-1 expression and that p53 can induce endogenous Egr-1 expression also, whereas mutant p53 cannot yield this result. U87MG cells were transfected with the APP promoter and treated or not treated with suramin. Forty-eight hours after transfection, the whole-cell protein was extracted for luciferase assay, and the results demonstrated that suramin can suppress APP promoter activities.
Physical interaction between Purα and Egr-1
Because Purα can suppress the up-regulatory effects of Egr-1 on APP promoter activities and inhibit the endogenous expression of the Egr-1 gene, the physical interaction between these two transcriptional factors was subsequently investigated. We designed a competition EMSA to analyze the effects of the binding of the two TFs to the APP promoter. The results demonstrated that both Egr-1 and Purα can bind to the APP promoter 5’-UTR; however, an increase in the amount of Egr-1 protein resulted in the disappearance of Purα-binding bands and its replacement by Egr-1-binding bands (Figure 9A). In contrast, an increase in the amount of Purα protein resulted in the disappearance of Egr-1- binding bands and its replacement by Purα-binding bands (Figure 9B). These results implied that Egr-1 and Purα can completely bind to the APP promoter. Further CoIP and pull-down assays were performed to assess the physical interaction between the two TFs. Forty-eight hours after U87MG cells were co-transfected with Purα and Egr-1 eukaryotic expression plasmids, the cells were harvested, and the cellular proteins were extracted for CoIP. The results showed that the two proteins did not physically interact (data not shown). To further confirm the physical interaction between the two proteins, a pull-down assay was performed with GSH-Purα and Egr-1 protein or with GSH-Egr-1 and Purα protein in vitro, and no physical interaction was observed in either case (data not shown). These results implied that the two proteins may only competitively bind to the binding sites in the 5’-UTR of the APP promoter to exert regulatory effects on APP gene expression, but that there were no physical interactions between these two proteins. Some other mechanisms may exist, and we hypothesize that the two TFs exhibit displacement, i.e., one protein can displace the other to affect its functions (Figure 10). To further investigate the distributions of these two transcriptional factors within the cells, the EMFs were used for immunohistochemistry examination. The results demonstrated that the two proteins are located in both the cytoplasm and nucleus, and colocalization could be observed at both sites (Figure 9C).
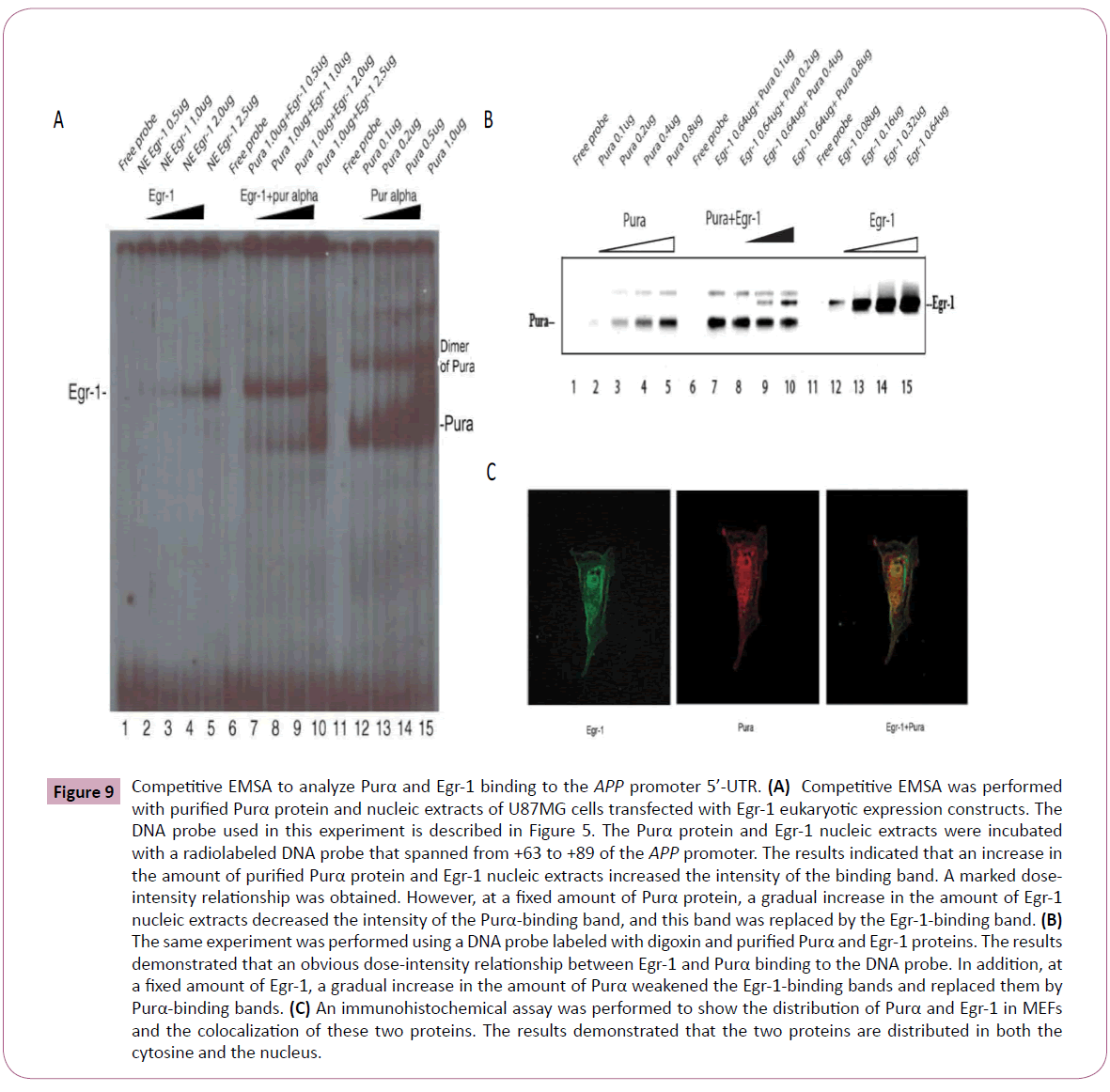
Figure 9: Competitive EMSA to analyze Purα and Egr-1 binding to the APP promoter 5’-UTR. (A) Competitive EMSA was performed with purified Purα protein and nucleic extracts of U87MG cells transfected with Egr-1 eukaryotic expression constructs. The DNA probe used in this experiment is described in Figure 5. The Purα protein and Egr-1 nucleic extracts were incubated with a radiolabeled DNA probe that spanned from +63 to +89 of the APP promoter. The results indicated that an increase in the amount of purified Purα protein and Egr-1 nucleic extracts increased the intensity of the binding band. A marked doseintensity relationship was obtained. However, at a fixed amount of Purα protein, a gradual increase in the amount of Egr-1 nucleic extracts decreased the intensity of the Purα-binding band, and this band was replaced by the Egr-1-binding band. (B) The same experiment was performed using a DNA probe labeled with digoxin and purified Purα and Egr-1 proteins. The results demonstrated that an obvious dose-intensity relationship between Egr-1 and Purα binding to the DNA probe. In addition, at a fixed amount of Egr-1, a gradual increase in the amount of Purα weakened the Egr-1-binding bands and replaced them by Purα-binding bands. (C) An immunohistochemical assay was performed to show the distribution of Purα and Egr-1 in MEFs and the colocalization of these two proteins. The results demonstrated that the two proteins are distributed in both the cytosine and the nucleus.
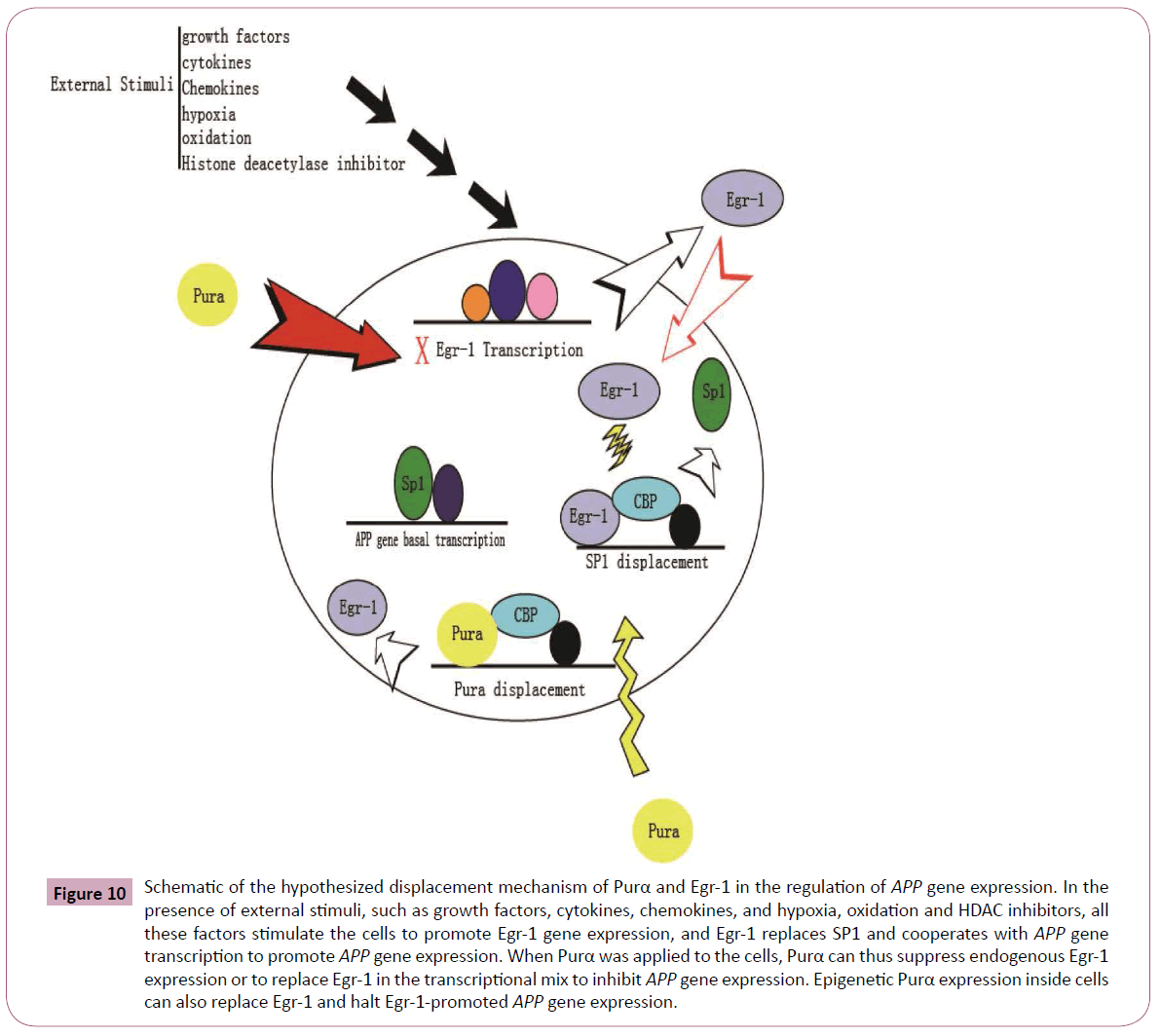
Figure 10: Schematic of the hypothesized displacement mechanism of Purα and Egr-1 in the regulation of APP gene expression. In the presence of external stimuli, such as growth factors, cytokines, chemokines, and hypoxia, oxidation and HDAC inhibitors, all these factors stimulate the cells to promote Egr-1 gene expression, and Egr-1 replaces SP1 and cooperates with APP gene transcription to promote APP gene expression. When Purα was applied to the cells, Purα can thus suppress endogenous Egr-1 expression or to replace Egr-1 in the transcriptional mix to inhibit APP gene expression. Epigenetic Purα expression inside cells can also replace Egr-1 and halt Egr-1-promoted APP gene expression.
Discussion
The results obtained in this study provide the first demonstration of the mechanisms through which Purα and Egr-1 regulate APP gene expression and of the mechanism through which the interactions between these two transcriptional factors regulate APP gene expression. Based on the experimental results obtained in this study, several novel observations have been put forward for consideration of the mechanisms responsible for the regulation of APP gene expression. There are several binding sites in the proximal end and 5’-UTR of the APP promoter, the DNA sequence of which is also characterized by GC-rich nucleotides. The transcriptional factors Purα and Egr-1 preferentially bind to GC-rich regions, and there are two Egr-1-binding sites (+63 to +67 and +66 to +82) and one Purα-binding site (+63 to +67) were discovered in the 5’-UTR of the APP promoter. All of these sites control the regulatory effects of these two TFs on APP promoter activities. The close location and overlapping position will spatiotemporally interfere with the binding of the two TFs to these sites to exhibit there functions. These observations also support the hypothesis of a displacement mechanism underlying the functions of these two TFs.
Based on previous studies that have demonstrated that Purα can negatively regulate the APP promoter activity, the western blots and immunohistochemistry results also illustrate that the knockdown of Purα results in increases in the APP expression level in both cells and brain tissues. The EMSA and ChIP’s assay results also verified the existence of Purα-binding sites in the 5’- UTR of the APP promoter [27]. Considering the specificity of the Purα-protein binding site, we hypothesized that the mechanism underlying this negative regulation of Purα on APP gene expression was due to binding to this site, but further evidence of this regulation still needs to be investigated. Given the location and sequence homology within the APP promoter sequences, we extended our studies to include the Egr-1 protein, a zinc finger transcription factor, and investigated its effects in the presence and absence of Purα. The luciferase reporter assay results demonstrated that Egr-1 can upregulate APP gene expression.
Egr-1 is an important regulatory factor for the expression of many genes in the body. Hendrickx et al. reported that transcription of the Egr-1 gene to be regulated by APP. In primary cultures of cortical neurons, APP significantly down regulation Egr-1 expression at both mRNA and protein levels in a γ-secretase independent manner and that APP fosters a low level of Egr-1 and c-fos expression in the mouse prefrontal cortex by inhibiting CREB recruitment and improving HDAC2 recruitment to the corresponding gene promoters [33,34]. Koldamova et al. reported that genes associated with Egr-1 binding revealed a set of related networks including synaptic vesicle transport, clathron mediated endocytosis, intracellular membrane fusion and transmission of signals elicited by Ca2+ influx. Egr-1 binding is associated with significant enrichment of activating chromatin markers and appears enriched near genes that are up-regulated in the brains of APP mice [23]. Among the putative Egr-1 targets included those related to synaptic plasticity and transport of proteins, such as Arc,Grin1, Syn2, Vamp2 and Stx6 as well as genes implicated in AD such as Picalm,Psen2 and APP [35]. Egr-1 is closely associated with spatial learning, memory formation, and cognitive ability and considered as the memory-related early gene Egr-1 in the pathogenesis Alzheimer’s disease. Cognitive and cerebrovascular deficits are 2 landmarks of Alzheimer's disease (AD) to target for effective therapy. Papadopoulos P et al. reported that simvastatin failed to improve spatial learning and memory deficits and the decreased baseline levels of the memory-related protein early growth response-1 (Egr-1) in the hippocampus CA1 area [36]. It is obvious that Egr-1 is an important regulatory factor in the nervous system and that the relationship between Egr-1 and APP is also pivotal for memory formation in Alzheimer’s disease. Our results focused on the effects of Egr-1 on APP gene expression and illustrated that Egr-1 can upregulate APP gene expression. It is not hard to understand that there may be a regulatory loop between APP and Egr-1, but the detailed mechanism remains to be further investigated.
Purα is another highly conserved developmentally regulated protein that is ubiquitous in nature and plays a critical role in neuronal development and synaptogenesis. Purα is a multifunctional protein that binds to single-strand nucleic acids in a sequence-specific manner and is a member of a protein family that is strongly conserved from bacteria through human [25]. Purα has been most extensively characterized as a sequencespecific single-stranded DNA- and RNA-binding protein that was originally cloned based on its affinity for a single-stranded DNA element with the GGNGGN sequence [28,37]. Our previous study found that Purα can negatively regulate APP gene expression, but the detailed mechanism underlying this regulation has not been investigated. Based on our previous studies, we extended our current study to an investigation of Egr-1, another transcriptional factor that preferentially binds to the DNA sequence in a GCrich region to investigate the mechanisms through which Purα regulates APP gene expression. According to the analysis of the APP promoter sequence, particularly its 5’-UTR, we found a specific region that has both Purα- and Egr-1-binding sites.
Histone deacetylase (HDAC) inhibitors are also important factors for inducing Egr-1 reactivation and expression and also increase the transactivities for the expression of many genes [38,39]. These have been utilized as epigenetic modifiers for the treatment of a number of CNS disorders, including epilepsy, schizophrenia, and Alzheimer's diseases [40, 41]. It has been reported that the epigenetic regulation of immediate-early genes involved in memory formation, and one of the key signaling pathways under epigenetic control is the regulatory immediateearly gene (IEG) Egr-1. The 5’ cis-regulatory elements in the promoter of Egr-1 contains binding sites for several regulatory factors, including two cAMP response element (CRE) sites that bind CREB, six serum response elements (SRE) sites that bind ELK1, activating protein-1/2 (AP-1/2) sites that bind Fos/Jun dimers, an SP1 site, an CCAAT/enhancer binding protein (C/EBP) site, and GSG box sites that bind EGR-family members [42, 43]. The current study focused on the mechanism of Pura and Egr- 1 in regulation of APP promoter activities and HDAC inhibitors were used to stimulate endogenous Egr-1 expression and we found that Egr-1 is a positive regulator for APP promoter, Pura acted as a negative regulator for APP promoter, both of these two transcriptional factors competitively bind to the specific binding sites existed in the 5’-UTR of APP promoter in which they are close in the spatial and overlapped. In other hand, Pura suppressed the endogenous Egr-1 expression. Our results demonstrated that a HADC inhibitor can markedly increase both APP promoter activity and endogenous Egr-1 expression. We conclude that the reason for the increase in APP promoter activities is due to an increase in endogenous Egr-1 expression because Egr-1 can upregulate APP promoter activities. However, the transfection of cells with a Purα expression construct ameliorated the effects of both TSA and butyrate. In fact, Purα alone was able to reduce the basal promoter activity of APP, as has been reported previously [27]. Surprisingly, Purα also minimized the ability of HDAC inhibitors to activate APP gene expression. This is a novel finding, and the mechanisms underlying how Purα counteracts the effect of HDAC inhibitors requires further investigation of the endogenous Egr-1 expression level by western blotting. The results demonstrated that Egr-1 was activated by both butyrate and TSA. In parallel, TSA induced the binding of Egr-1 to the APP probe, as determined by EMSA. These data suggest that HDAC inhibitors increased endogenous Egr-1 expression and promote APP binding activity, but this does not result in an increase in APP expression in the presence of Purα because Purα strongly suppresses APP promoter activity, even when Egr-1 and Purα are co-transfected into HEK293 or U251MG cells, which indicates that Purα can eliminate the up-regulatory effects of Egr-1 on the APP promoter. A series of experiments were designed to verify the effects of changes in the endogenous Egr-1 levels on the APP promoter activities, and the Egr-1 inhibitor suramin and the Egr-1 activator p53 were used to examine the endogenous Egr-1 expression and its effect on APP promoter activities. The experimental results demonstrated that suramin can inhibit endogenous Egr-1 expression, even after treatment with TSA. In addition, we also found that suramin can suppress APP promoter activities and that p53 can increase endogenous Egr-1 expression when transfected into U87MG cells. We also observed that Purα can suppress endogenous Egr- 1 expression when transfected into U87MG cells. Above all, it is not difficult to conclude that one reason for the Purα-induced suppression of the APP promoter activities is its interplay with Egr-1. The detailed mechanism for this is unknown, and further investigations are needed to elucidate this mechanism.
To understand the importance in the change in Egr-1 levels and binding activity in the presence and absence of Purα, we correlated promoter activation with promoter binding intensity, which was measured by EMSA, and the total protein levels, which were measured by western blotting, to determine the relationship between the total protein levels and binding ability. We evaluated the direct binding between the Egr-1 and Purα proteins and mapped the regions of these proteins that are responsible for their functional activation and enhancement of nucleic acidbinding activity. The experimental results demonstrated that a competing binding relationship between the two transcriptional factors when they bind to the APP promoter. An increase in the amount of Egr-1 protein decreased the binding abilities of Purα, and vice versa. In addition, an increase in the amount of Purα protein also decreased the binding abilities of Egr-1 to the APP promoter. These phenomena indicate the possibility that a displacement mechanism is responsible for the effects of these two transcriptional factors in the regulation of APP promoter activities because they share overlapping binding sites in the 5’-UTR of the APP promoter. The in vitro physical interaction of the two TFs showed that they do not exhibit a direct physical interaction. We hypothesize that a displacement mechanism is responsible for the regulation of APP gene expression by these two TFs. In the presence of external stimuli, such as growth factors, cytokines, chemokines, hypoxia, oxidation and HDAC inhibitors, endogenous Egr-1 transcription was activated, and the resulting increased levels of Egr-1 displace SP1, as described by Khachigian [44] (Figure 11), to promote APP transcription. The introduction of Purα into the cells suppresses APP gene expression through two mechanisms: the first mechanism involves inhibiting Egr-1 transcription and reducing the endogenous level of Egr-1, and the second mechanism involves the competitive binding to the APP promoter and eliminating Egr-1 binding and thereby downregulating APP gene expression. This hypothesis is based on the current experimental results, but additional studies are required to provide detailed evidence supporting this hypothesis. These findings are correlated with the subcellular localization obtained through immunohistochemistry analysis. These studies provide information regarding whether subcellular localization affects the function and binding activity of these proteins.
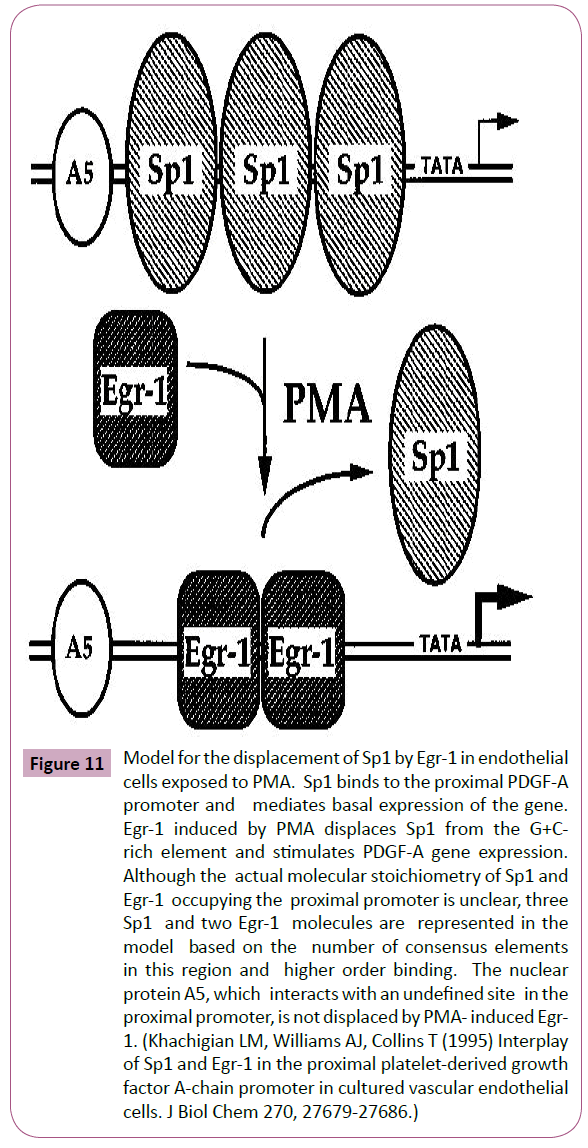
Figure 11: Model for the displacement of Sp1 by Egr-1 in endothelial cells exposed to PMA. Sp1 binds to the proximal PDGF-A promoter and mediates basal expression of the gene. Egr-1 induced by PMA displaces Sp1 from the G+Crich element and stimulates PDGF-A gene expression. Although the actual molecular stoichiometry of Sp1 and Egr-1 occupying the proximal promoter is unclear, three Sp1 and two Egr-1 molecules are represented in the model based on the number of consensus elements in this region and higher order binding. The nuclear protein A5, which interacts with an undefined site in the proximal promoter, is not displaced by PMA- induced Egr- 1. (Khachigian LM, Williams AJ, Collins T (1995) Interplay of Sp1 and Egr-1 in the proximal platelet-derived growth factor A-chain promoter in cultured vascular endothelial cells. J Biol Chem 270, 27679-27686.)
Conclusion
The mechanism of Purα on APP gene expression has been studied in the current research work. The luciferase assay has been employed to check the effects of Purα on APP gene expression. The research demonstrated that the binding sites of Purα and Egr-1 on 5’-UTR determined the regulatory effects of these two transcriptional factors. Since the binding sites close in the spatial position and also overlapped, binding of one transcriptional factors will affected the other’s binding. In this way the competitive binding existed and the displacement hypothesis was also promoted. Egr-1 behaved as a positive regulator and when the binding sites in 5’UTR have been deleted,both Egr-1 and Purα lose the regulatory effects on the APP gene expression. Purα acted as a down-regulator for endogenous Egr-1 expression. Luciferase results demonstrated that Purα can suppress Egr-1 promoter activities. The results of ChIP’s assay and EMSA confirmed the binding of the two TFs on APP promoter 5’-UTR and also confirmed the interaction between the Purα and Egr-1 on APP gene expression. To disturb the endogenous Egr-1 expression and competitively bind to the 5’-UTR of APP gene promoter, Purα exert its negative regulation on APP gene expression. As human transcriptional activating factors, the negative regulation of Purα on APP gene expression possesses a great importance in AD pathogenesis and might be open a new insight for the prevention and treatment of AD in the future. The detailed mechanisms between these two TFs need further deep investigations.
Acknowledgment
We thank Drs. Jennifer Gorden and Kamel Khalili for providing Pur-α-knockout MEFs. We also thank the past and current postgraduate students of the Ningxia Key Laboratory of Cerebrocranial Diseases for their valuable work.
This work was supported by grants from NFCS to Dr. Jianqi Cui (81260197) and the National 973 Project for Dr. Tao Sun (2012CB722408) and Dr. Jianguo Niu (2014CB560711)
6901
References
- Lahiri DK, Maloney B (2010) The "LEARn" (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer's disease, and proposes remedial steps.ExpGerontol 45: 291-296.
- Agca C, Fritz JJ, Walker LC, Levey AI, Chan AW, et al. (2008) Development of transgenic rats producing human beta-amyloid precursor protein as a model for Alzheimer's disease: transgene and endogenous APP genes are regulated tissue-specifically.BMC Neurosci 9: 28.
- Takahashi K, Niidome T, Akaike A, Kihara T, Sugimoto H (2009) Amyloid precursor protein promotes endoplasmic reticulum stress-induced cell death via C/EBP homologous protein-mediated pathway. J Neurochem 109: 1324-1337.
- Mattson MP (1997) Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 77: 1081-1132.
- Lee S, Das HK (2010) Transcriptional regulation of the presenilin-1 gene controls gamma-secretase activity.Front Biosci (Elite Ed) 2: 22-35.
- Lin HK, van der Wel PC (2014) How amyloid precursor protein protects itself from cleavage. Structure 22: 361-362.
- Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, et al. (1989) Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease.N Engl J Med 320: 1446-1452.
- Johnson SA, McNeill T, Cordell B, Finch CE (1990) Relation of neuronal APP-751/APP-695 mRNA ratio and neuritic plaque density in Alzheimer's disease.Science 248: 854-857.
- Ohta M, Kitamoto T, Iwaki T, Ohgami T, Fukui M, et al. (1993) Immunohistochemical distribution of amyloid precursor protein during normal rat development.Brain Res Dev Brain Res 75: 151-161.
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, et al. (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity.Cell 81: 525-531.
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, et al. (2009) Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription.ProcNatlAcadSci U S A 106: 3907-3912.
- Ohyagi Y, Tabira T (1993) Effect of growth factors and cytokines on expression of amyloid beta protein precursor mRNAs in cultured neural cells. Brain Res Mol Brain Res 18: 127-132.
- Trejo J, Massamiri T, Deng T, Dewji NN, Bayney RM, et al. (1994) A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer's beta-amyloid precursor protein gene. J BiolChem 269: 21682-21690.
- Yoshikai S, Sasaki H, Doh-ura K, Furuya H, Sakaki Y (1990) Genomic organization of the human amyloid beta-protein precursor gene. Gene 87: 257-263.
- Belandia B, Latasa MJ, Villa A, Pascual A (1998) Thyroid hormone negatively regulates the transcriptional activity of the beta-amyloid precursor protein gene. J BiolChem 273: 30366-30371.
- König G, Masters CL, Beyreuther K (1990) Retinoic acid induced differentiated neuroblastoma cells show increased expression of the beta A4 amyloid gene of Alzheimer's disease and an altered splicing pattern. FEBS Lett 269: 305-310.
- Yu HT, Chan WW, Chai KH, Lee CW, Chang RC, et al. (2010) Transcriptional regulation of human FE6, a ligand of Alzheimer's disease amyloid precursor protein, by Sp1. J Cell Biochem 109: 782-793.
- Cuesta A, Zambrano A, Royo M, Pascual A (2009) Thetumour suppressor p53 regulates the expression of amyloid precursor protein (APP). Biochem J 418: 643-650.
- Wang PL, Niidome T, Akaike A, Kihara T, Sugimoto H (2009) Rac1 inhibition negatively regulates transcriptional activity of the amyloid precursor protein gene. J Neurosci Res 87: 2105-2114.
- Wen Y, Yu WH, Maloney B, Bailey J, Ma J, et al. (2008) Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing.Neuron 57: 680-690.
- Gashler A, Sukhatme VP (1995) Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors.Prog Nucleic Acid Res MolBiol 50: 191-224.
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, et al. (2001) A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories.Nat Neurosci 4: 289-296.
- Renbaum P, Beeri R, Gabai E, Amiel M, Gal M, et al. (2003) Egr-1 upregulates the Alzheimer's disease presenilin-2 gene in neuronal cells.Gene 318: 113-124.
- Lu Y, Li T, Qureshi HY, Han D, Paudel HK (2011) Early growth response 1 (Egr-1) regulates phosphorylation of microtubule-associated protein tau in mammalian brain. J BiolChem 286: 20569-20581.
- Johnson EM (2003) The Pur protein family: clues to function from recent studies on cancer and AIDS.Anticancer Res 23: 2093-2100.
- Khalili K, Del Valle L, Muralidharan V, Gault WJ, Darbinian N, et al. (2003) Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse.Mol Cell Biol 23: 6857-6875.
- Darbinian N, Cui J, Basile A, Del Valle L, Otte J, et al. (2008) Negative regulation of AbetaPP gene expression by pur-alpha.J Alzheimers Dis 15: 71-82.
- Bergemann AD, Johnson EM (1992) The HeLaPur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication.Mol Cell Biol 12: 1257-1265.
- Hoffman PW, Chernak JM (1994) The rat amyloid precursor protein promoter contains two DNA regulatory elements which influence high level gene expression.BiochemBiophys Res Commun 201: 610-617.
- Izumi R, Yamada T, Yoshikai S, Sasaki H, Hattori M, et al. (1992) Positive and negative regulatory elements for the expression of the Alzheimer's disease amyloid precursor-encoding gene in mouse.Gene 112: 189-195.
- La Fauci G, Lahiri DK, Salton SR, Robakis NK (1989) Characterization of the 5'-end region and the first two exons of the beta-protein precursor gene.BiochemBiophys Res Commun 159: 297-304.
- Lahiri DK, Ghosh C, Ge YW (2003) A proximal gene promoter region for the beta-amyloid precursor protein provides a link between development, apoptosis, and Alzheimer's disease.Ann N Y AcadSci 1010: 643-647.
- Hendrickx A, Pierrot N, Tasiaux B, Schakman O, Brion JP, et al. (2013) Epigenetic induction of EGR-1 expression by the amyloid precursor protein during exposure to novelty.PLoS One 8: e74305.
- Hendrickx A, Pierrot N, Tasiaux B, Schakman O, Kienlen-Campard P, et al. (2014) Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease.PLoS One 9: e99467.
- Koldamova R, Schug J, Lefterova M, Cronican AA, Fitz NF, et al. (2014) Genome-wide approaches reveal EGR1-controlled regulatory networks associated with neurodegeneration.Neurobiol Dis 63: 107-114.
- Papadopoulos P, Tong XK, Hamel E (2014) Selective benefits of simvastatin in bitransgenicAPPSwe,Ind/TGF-β1 mice.Neurobiol Aging 35: 203-212.
- Bergemann AD, Ma ZW, Johnson EM (1992) Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein.Mol Cell Biol 12: 5673-5682.
- Su L, Cheng H, Sampaio AV, Nielsen TO, Underhill TM (2010) EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor.Oncogene 29: 4352-4361.
- Shimoyamada H, Yazawa T, Sato H, Okudela K, Ishii J, et al. (2010) Early growth response-1 induces and enhances vascular endothelial growth factor-A expression in lung cancer cells.Am J Pathol 177: 70-83.
- Urdinguio RG, Sanchez-Mut JV, Esteller M (2009) Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies.Lancet Neurol 8: 1056-1072.
- Haggarty SJ, Tsai LH (2011) Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity.Neurobiol Learn Mem 96: 41-52.
- Schwachtgen JL, Campbell CJ, Braddock M (2000) Full promoter sequence of human early growth response factor-1 (Egr-1): demonstration of a fifth functional serum response element.DNA Seq 10: 429-432.
- Sakamoto KM, Bardeleben C, Yates KE, Raines MA, Golde DW, et al. (1991) 5' upstream sequence and genomic structure of the human primary response gene, EGR-1/TIS8.Oncogene 6: 867-871.
- Khachigian LM, Williams AJ, Collins T (1995) Interplay of Sp1 and Egr-1 in the proximal platelet-derived growth factor A-chain promoter in cultured vascular endothelial cells.J BiolChem 270: 27679-27686.