Keywords
Depression; Chronic inflammation; Chronic renal failure; Systemic arterial hypertension; Diabetes mellitus
Introduction
According to the Global Burden of Disease Study 2015 [1], cerebrovascular disease (CVD) represents the second cause of years of life lost and one of the major causes of disability. This disability includes not only physical limitations and functional impairment, but also cognitive issues having substantial impact on the patient’s quality of life. Latin America is not excluded from the worldwide population aging process; this explains the increase in cardiovascular risk factors and, consequently, higher probability of occurrence of CVD.
There has been major progress in the understanding of cognitive impairment related to vascular pathology. These advances have made it possible to better define vascular cognitive impairment (VCI), and to improve our knowledge of the pathophysiology according to the type of vessel affected (large or small). Also, the wider use of cognitive screening tests for the detection of VCI, even in the early stages, has made it possible to establish the diagnosis in preclinical stages, with the purpose of controlling risk factors.
The Worldwide Report on Alzheimer’s Disease 2015 estimates that there are 9.2 million cases of dementia in Latin America, with a prevalence of 8.2% [2]. Certainly, the most frequent cause of dementia is Alzheimer’s disease (AD), followed by vascular dementia (VD) with 15% of dementia cases [3]. There are no data by country in Latin America.
The main objective of this consensus was to analyze and summarize current evidence on definition, classification, pathophysiology, diagnosis and treatment of vascular cognitive impairment from a Latin America perspective by a group of Hispanic specialists.
Epidemiology
Stroke is a leading cause of cognitive impairment, disability and mortality in Latin America, although information regarding stroke and vascular dementia in the region is limited [4-7]. Incidence rates of stroke reported in Latin American studies (all adjusted for Segi’s world population) range from 76.5 to 110 first-ever strokes per 100 000 per year [8-11]. Stroke prevalence per 1000 population, based on door-to-door surveys, ranges from 1.7 in rural areas to 8.7 among predominantly urban population. In older patients, (aged ≥60), prevalence of stroke ranges from 18.2 to 46.7 per 1000 [12-14]. Considering the high incidence and prevalence of stroke in Latin America, cerebral vascular injury should be expected to be a common cause of dementia and of milder forms of cognitive dysfunction. The prevalence of dementia in Latin America is comparable to that reported in North America. A review of 8 studies on dementia from 6 Latin American countries (Uruguay, Chile, Brazil, Venezuela, Cuba, Peru) reported an overall prevalence of 7.1% (with AD as the most frequent cause), without describing vascular etiology [15]. A report on the global epidemiology of dementia in developed and underdeveloped countries, reported a prevalence of 1.9% (1.0-3.0) in Cuba, 0.9% (0.06-1.78) and 2.2% (1.6-2.7) in Brazil [16]. In Mexico, a study of 110 patients with first-ever stroke, reported a prevalence of vascular dementia (three months after the CVA) of 12%, with a higher prevalence in illiterate patients and lower education [17].
Given the fast and increasing knowledge in this field, it is important to obtain accurate up-to-date data. For this reason, a group of experts in cerebrovascular pathology, including neurologists, geriatricians and psychiatrists from several Latin American countries, analyzed the information available in this region, as well as worldwide data regarding cognitive impairment of vascular etiology. The goal of the study group was to establish definitions, diagnosis, classification and recommendations for the prevention and treatment of vascular cognitive impairment (VCI).
Methodology for Study Group’s Recommendations
A meeting was held on April 2016 in Miami, Florida, USA, including a group of Hispanic specialists (vascular neurologists, geriatrics and psychiatrists) of the following countries: Mexico, Guatemala, El Salvador, Costa Rica, Peru, Bolivia, Chile, Paraguay, Colombia, Panama, Venezuela, Dominican Republic, Honduras and Nicaragua. Likewise, Hispanic experts in vascular dementia (Spain, Italy and United States) also participated in the consensus.
The Delphi [18] method was used by the experts, who reviewed the information available (provided months before the meeting) related to assigned topics: definition of VCI, risk factors, pathophysiology, neuropsychological and imaging diagnosis, and treatment. The Grade [19] System was used for assigning the evidence and recommendation of available treatments. The discussion panels analyzed and discussed the available evidence on VCI for proposed definitions, risk factors and diagnostic items. Subsequently, the conclusions of each panel were written, and study groups reviewed them until a global consensus was reached. Once this process was completed, the preparation of the final document was carried out.
Definition from mild vascular cognitive impairment to vascular dementia
In 1988, Reisberg et al. [20] introduced the Global Deterioration Scale (GDS) in which stage GDS 3 indicated the mild progression of cognitive impairment. It was not until 1999, however, when Petersen et al. [21] consolidated the concept of mild cognitive impairment (MCI). MCI emerges then as a prodromal stage of AD, characterized as an interim cognitive stage between normal cognition and dementia. Since then, it is established that to cross the borderline between MCI and dementia, the grade of cognitive dysfunction should be one that impacts on functionality. Therefore, investigations were conducted in this regard, other causes of MCI additionally to AD were recognized, a sub-classification depending on the combination of involved cognitive domains was created, and an attempt was made to associate it with a specific cause (including the vascular cause) as the following step in the diagnostic process [22].
The expansion of spectrum of cognitive impairment with a cerebrovascular origin arises in this way, not only in terms of etiology but also for the severity. Several terms are used to refer to prodromal stages of vascular dementia: mild vascular cognitive impairment (M-VCI), preclinical vascular cognitive impairment, vascular pre-dementia, and non-dementia vascular cognitive impairment [23], referring basically to the same condition but with small differences in the operational definition of the term conferring a great heterogeneity about information of this entity. In 2009, the first special symposium was held at the fourth edition of the Congress of International Society for Vascular Behavioral and Cognitive Disorders (VASCOG), in which the term vascular cognitive impairment (VCI) is proposed to name any cognitive dysfunction with a vascular cause, regardless of its specific etiology and grade of severity, sub-dividing this term into Major Vascular Cognitive Impairment (equivalent to vascular dementia) and Mild Vascular Cognitive Impairment (similar to VCI). In this way, the spectrum becomes more complete in all its dimensions and the prodromal stage of dementia with a vascular cause is better delimited under the name Mild Vascular Cognitive Impairment [23]. On the other hand, the American Psychiatric Association, in DSM-5, calls it minor neurocognitive disorder [24]. The criterion differentiating a minor from a major disorder is that cognitive difficulties should not influence a person’s capability to perform activities of daily life (ADL). It is considered a major neurocognitive disorder when it affects ADL.
The term “cognitive impairment” is classically used for this entity, while the term “neurocognitive” used in DSM-5 emphasizes a condition characterized by cognitive failures not caused by a psychiatric disorder, such as major depression or schizophrenia, but resulting from an “organic” etiology [24]. Thus, VCI is beginning to be recognized as encompassing many diseases, each with different severity and impairment patterns. The recognition that a decline in prior cognitive ability has occurred, as documented by longitudinal data or inferred from a premorbid reference, is implicit in the term.
VASCOG recognizes that there are two requirements for diagnosis of a cognitive disorder: a subjective report and objective evidences of alterations. A clinical visit for a cognitive disorder as the reason for consultation usually results from a concern by the patient and/or an informant who has observed a decrease in the cognitive function. In vascular dementia or major VCI, the subjective report will typically be that the individual depends on other to plan or make decisions, has had to leave complex projects, repeats the same conversation, needs reminders to perform a task, has significant difficulties with expressive or receptive language, has problems to navigate a familiar environment, or has clear alteration in body schema, capability to calculate, read or write [23].
Recommendations
The concept of VCI is a construct covering the entire spectrum of cognitive disorders ranging from mild cognitive impairment (MCI) through fully developed dementia, due to all forms of vascular brain injury. It includes vascular disease as a single etiology, but also in combination with other causes of cognitive impairment, mainly neurodegenerative disorders. Two requirements for diagnosis of a cognitive disorder include a subjective report and objective evidence of cognitive impairment. The criterion to distinguish a minor from a major disorder is that cognitive difficulties do not influence the subject’s capability to perform ADLs, whereas these are involved in a major neurocognitive disorder.
Diagnostic Criteria for Vascular Cognitive Impairment
The most commonly used criteria for the diagnosis of vascular dementia are those stated by the National Institute of Neurological Disorders and Stroke and the Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) [25], those adopted by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers (ADDTC) [26], and those contained in DSM-5 and CIE-10. In the last meeting of the VASCOG in 2013, it was recognized that cognitive disorders of vascular etiology are a heterogeneous set of alterations with different etiologies and clinical manifestations, all of which are considered under the term “vascular cognitive disorders” [23]. There are two critical areas for the diagnosis of vascular dementia: one, the certainty of the presence of a cognitive disorder, and two, the determination that vascular disease is the dominant, if not the only pathology, which accounts for the cognitive disorder.
Clinical characteristics of cognitive syndrome
To clinically establish a predominantly vascular etiology for a cognitive disorder, VASCOG recommends that the following characteristics should be considered in addition to the subjective report and objective evidences of cognitive alterations. The alteration of executive functions, unlike memory disturbances, is often the most outstanding feature of vascular dementia. Memory impairment may not be present in some cases; in others, the disorder in episodic memory is typical (although not exclusive) of vascular dementia. The heterogeneity of pathology in VCI suggests that cognitive deficits will vary according to involved brain area and the way in which lesions appear. This pattern is time-related to strokes, hemorrhages or other vasculopathies [23,24]. However, sometimes it is difficult to establish this temporal relation clinically. For example, cognitive impairment is at its highest level shortly after a brain hemorrhage and it may show a significant improvement over the next three months; but persistence beyond this period is considered necessary for the diagnosis of cognitive impairment. VCI with a gradual onset and slow progression is usually due to small-vessel disease leading to lesions in the white matter, basal ganglia, and/or thalamus [25]. This gradual progression is often marked by acute events that leave subtle neurological deficits such as focal weakness, unilateral incoordination, asymmetrical reflexes, instability, short-step gait, or signs of Parkinsonism [26]. Cognitive changes may be attributed to interruption of cortico-subcortical circuits, and it is likely that speed of information processing, complex attention and executive functions may be involved. White matter ischemic lesions are commonly associated with frontal executive deficits, regardless of their distribution in the brain [27]. Vascular lesions may disrupt thalamus-cortical, striatal-cortical and basal ganglia-prefrontal cortex pathways, as well as cortical and limbic structures; therefore, VCI is often associated with behavioral and emotional disturbances. As these neuropsychiatric features are not specific to vascular etiology, they are not considered as core characteristics in the diagnosis [28]. A clinical history and an appropriate neurological examination may provide additional information, or may be the only objective source of evidence, in the absence of neuroimaging. Thus, a well-documented history of acute stroke is solid evidence of cerebrovascular disease, either from large-vessel disease, or from embolism. Additionally, evidence is also provided by neurological examination showing typical stroke signs, including hemiparesis, facial asymmetry, sensory disorder including visual field defects and pseudobulbar syndrome (supranuclear weakness in facial muscles, tongue and pharynx, spastic dysarthria, difficult swallowing and loss of emotional control) [23]. The following criteria support the presence of CVD, but are not enough by themselves to establish vascular disease as a probable cause of VCI:
1. Early presence of a gait disorder.
2. Urinary urgency or incontinence and other urinary symptoms not explained by urological or non-cognitive neurological disease.
3. Changes in personality and mood, abulia, depression, emotional incontinence, or other subcortical deficits, including psychomotor retardation and abnormal executive function [23,25,26].
Determination of significant cerebrovascular disease (CVD)
This part of the diagnostic process is based on clinical history and neuroimaging. The demonstration of vascular lesions in neuroimaging is essential for higher diagnostic certainty. Absence of neuroimaging data may lead to inaccurate diagnosis. Neuroimaging is also important to rule out less common causes, such as a brain tumor or the normal pressure hydrocephalus (NPH) syndrome [29]. In addition, imaging is important in determining the vascular contribution to AD or to frontotemporal degeneration, as a mixed etiology of cognitive impairment [30]. In general, the evidence of significant vascular pathology is based on computed tomography (CT) or magnetic resonance imaging (MRI), the latter being the most sensitive for this purpose. Neuroimaging findings should be interpreted in the clinical context and their nature, severity and location must be determined. Attempts have been made to define the number of vascular lesions required to support a vascular etiology of cognitive impairment (see section 9.3.1 below, Neuroimagingsupported criteria) [31,32].
Two levels of certainty are recommended for a clinical VCI diagnosis: “probable” and “possible”. For a “probable” VCI diagnosis, both clinical and neuroimaging criteria should be met. Although rare, evidence of a genetic cerebrovascular disorder will support a “probable” level of certainty [33,34]. Examples include cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL); cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL); hereditary endotheliopathy with retinopathy, nephropathy and stroke (HERNS); pontine autosomal dominant microangiopathy and leukoencephalopathy (PADMAL); retinal vasculopathy with cerebral leukodystrophy (RVCL), and type IV A1 collagen disorder (e.g., mutations of COL4A1 gene) as well as familial forms of cerebral amyloid angiopathy (CAA) [35]. If clinical criteria are met but neuroimaging is not available, the certainty level of the VCI diagnosis remains “possible.” By obtaining cerebral imaging, the diagnosis progresses to a greater certainty level (“probable” VCI). The definitive level of diagnosis certainty is obtained through postmortem neuropathological examination. However, the term “definitive” VCI is not proposed in VASCOG recommendations as these recommendations are intended to establish clinical criteria [23].
Differential diagnosis
Given that incidental strokes and vascular leukoencephalopathy are common in the brain of elderly people, it is important to consider other possible etiologies when the patient’s cognitive impairment is assessed. When a non-vascular etiology of the dementia is provided by clinical history, physical examination, and/or laboratory investigations then VCI should not be the primary diagnosis but a contributing factor. Often, VCI is an important contributor to several neurodegenerative etiologies of dementia.
A history of early-onset memory deficits and progressive worsening of memory or language (transcortical sensory aphasia), motor skills (apraxia) and perception (agnosia), in the absence of corresponding focal lesions in brain imaging, are more suggestive of AD as the primary diagnosis. Likewise, VCI should not be diagnosed in the presence of other diseases that may induce cognitive impairment, such as a cerebral neoplasia, multiple sclerosis, encephalitis, toxic or metabolic causes. If the patient has major depression that is temporarily related to the onset of cognitive impairment, VCI should not be diagnosed [23]. Figure 1 depicts the diagnostic algorithm for vascular cognitive impairment proposed by VASCOG.

Figure 1: Diagnostic algorithm for vascular cognitive impairment.
Recommendations
Diagnosis of mild VCI or vascular dementia should be considered in patients with cognitive impairment and evidence of cerebrovascular lesions. Executive function abnormalities, rather than memory loss, are often the most outstanding feature of VCI. Memory impairment may not be present in some cases of VCI; in others, executive dysfunction with loss-of-set may cause memory problems resulting in episodic memory loss (typical but not exclusive of VCI). The heterogeneity of pathology in VCI suggests that cognitive deficits will vary according to involved brain area and the progression of lesions. When above cognitive profile is temporarily related to a history of ischemic or hemorrhagic strokes the diagnosis of VCI should be confirmed by neuroimaging studies.
Anatomical Subtypes of Vascular Cognitive Impairment
Anatomically, VCI may be classified as cortical, subcortical or cortico-subcortical considering the site of the predominant location of vascular damage. VCI may also be sub-categorized as ischemic or hemorrhagic, according to the main etiological type of CVD.
Cortical
It is uncommon to find pure cortical VCI, because vascular lesions involving exclusively cortical areas are rare; therefore, they are usually included in the cortico-subcortical group.
Subcortical
Vascular lesions may affect exclusively subcortical regions causing VCI. Several attempts have been made to characterize subcortical VCI. The pathological basis of subcortical VCI includes:
1. Multiple lacunar infarctions [26].
2. White matter lesions of vascular origin (Binswanger’s disease type) [27].
3. Thalamic dementia due to infarcts located in the thalamus [28].
Cortico-subcortical
Most cases of VCI consist of a combination of cortical and subcortical lesions [29]. In addition, VCI is subdivided into ischemic or hemorrhagic VCI, according to the major etiological type of CVD.
Etiology of Vascular Cognitive Impairment
The increasing recognition of cognitive impairment as a relevant consequence of CVD (beyond classically recognized alterations in motor, sensitive, and other functions) has led to a change in the approach and definition of CVD. Now, there is an appeal to understand the concept of cerebrovascular disease as a construct that may be associated or not with a phenotype of clinical expression for different diseases, according to location of damage, repair capability, previous substrate (prior cognitive threshold, interconnection networks, neuroplasticity), as well as inflammatory and regenerative processes characteristic of the host. Currently, it is considered that there may be “silent” (or rather clinically very subtle) lesions, which beyond the apparent absence of clinical expression, are relevant for cognitive function, and may be an indication of a pathophysiological process that requires treatment.
Parenchymal lesions
Evolution of knowledge on the histo-pathological origin of vascular dementia: Recent progress in histopathology has led to the emergence of new concepts of cerebrovascular disease relevant to cognition. The dimensions and location of lesions are relevant parameters for VCI expression:
Size of lesion: It is recognized that VCI may occur with a volume of damaged tissue of at least 100 mm3 regardless the location, or with a lower volume depending on location [30]. Thus, the concept of a strategically-localized infarct arises, in which a lower volume may have a greater cognitive impact due to its location [31,32]. However, the number of infarcts and the volume of damaged tissue correlate with dementia degree (mild = 3 cm3, moderate = 29 cm3, severe = 63 cm3) [33].
Location of lesion: Beyond the volume of damaged tissue, there are cerebral structures that because of their modular function performing a cognitive task or their importance as pathways connecting two cortical areas relevant for cognitive function may cause VCI after relatively small lesions. The following areas are recognized as strategic lesion sites [28,29,34,36,37]:
1. Limbic, paralimbic, frontal and parietal associative heteromodal cortex.
2. Subcortical structures such as basal forebrain, amygdala, (anterior, paramedian) thalamus, basal nuclei.
3. Papez memory circuit.
4. Frontal-subcortical circuits. In particular, prefrontalcaudate nucleus/thalamus/prefrontal circuits.
5. White matter substance (subcortical, deep).
6. Cortico-cortical association fibers.
7. Cortico-subcortical fibers.
8. Commissural fibers (corpus callosum, anterior commissure).
9. Lesions due to disconnection of the previous mentioned structures.
Types of vascular lesion causing cognitive impairment
In the classical concept, major or minor vascular cognitive impairment may be secondary to lesion in small vessels, large vessels or an association of both.
Lesions in large vessels: They are most commonly caused by an atherothrombosis or cardioembolism with occlusion of large cerebral arteries, either extracranial (mainly carotid arteries) or affecting the main intracranial arteries (middle cerebral, anterior cerebral and posterior cerebral arteries). This obstruction causes middle or large-sized strokes causing signs of a neurological disconnection decoupling modules required for the cognitive process. It may also occur due to the presence of multiple infarcts in different locations, which cause a great cortical dysfunction and a loss of different basic functions for cognition (language, memory, executive function, etc.).
Lesions in small vessels: These are classically considered secondary to lipohyalinosis, thickening of arteriolar walls (arteriolosclerosis), and occlusion of perforating arteries causing a small infarct area (microinfarcts, lacunes), usually in subcortical white matter areas or basal ganglia. It has also been described associated with microatheromatosis in the origin of perforating arteries. Small vessel disease is a major risk factor for developing cognitive impairment and dementia. The following types of small cerebral vessel lesions are recognized:
1. Lacunar infarct (lacune): There is no a consensus about the number and location of lacunes required for VCI diagnosis. Possibly, 1 or 2 lacunes do not cause cognitive impairment in older people, and they could be incidental findings. More than two lacunes should be considered necessary to support the VCI diagnosis, except for single lacunes in strategic areas.
2. Hyperintensity of the white matter: It is associated with silent neurological and cognitive symptoms.
3. Perivascular space: The general enlargement of perivascular spaces is associated with white matter hyperintensity and lacunar infarcts. Some studies have associated the most prominent perivascular spaces with a worse cognitive function.
4. Microbleeds: Lobular microbleeds have been included in the research criteria for cerebral amyloid angiopathy. New studies show an association between microbleeds and cognitive impairment.
In the definition of VCI neuropathological substrates, the Newcastle classification considers the following vascular alterations [35]:
1. Large infarcts or several infarcts: A result from vascular damage >50 mL of infarcted brain tissue. They include multi-infarct dementia.
2. Lacunar infarcts or multiple microinfarcts: Those leaving gliosis and small areas of cavitation. At least 3 infarcts with a minimum size of 0.5 mm are thought to be necessary. This item includes microbleeds, cerebral amyloid angiopathy and diffuse leukoencephalopathy.
3. Strategic infarcts: They are present in regions of thalamus and hippocampus, with a lower volume but a high impact on cognitive function.
4. Cerebral hypoperfusion with hippocampal sclerosis, ischemic anoxic damage, laminar cortical necrosis, or watershed infarcts: In older subjects, hippocampal sclerosis is accompanied by TDP-43 protein deposits.
5. Brain hemorrhages: either intracerebral (mainly lobar) or subarachnoid.
6. Mixed dementia: Presence of cerebrovascular changes with AD pathology.
On the other hand, Jellinger summarizes the morphological lesions causing vascular cognitive impairment as describe in Table 1 [38].
| Multifocal Disease |
Large vessels (large arteries dementia) |
Dementia caused by multiple infarcts: |
| Multiple infarcts |
| Watershed infarcts |
| Small vessels (small vessels dementia) |
Due to subcortical infarcts: |
| Multiple lacunar infarcts with peripheral lesions in the white matter |
| Granular atrophy of cortex (multifocal cortical microinfarcts) |
| Lacunar infarcts and multilacunar state |
| Binswanger’s subcortical leukoencephalopathy |
| Subcortical angiopathies (CADASIL and others) |
| Due to subcortical plus cortical affection: small multiple infarcts caused by: |
| Hypertensive or atherosclerotic angiopathy |
| Cerebral amyloid angiopathy with or without hemorrhage |
| Collagen or vascular inflammatory disease (systemic or primary vasculitis of nervous system, fibromuscular dysplasia) |
| Hereditary forms of cerebral amyloid angiopathy |
| Due to hypoperfusion or hypoxia/ischemia |
Incomplete white matter infarcts |
| Ischemia related to antiphospholipid syndrome |
| Hypoxic-ischemic encephalopathy (cortical laminar necrosis, post cardiac arrest, hypotension) |
| Associated with venous infarcts |
Large hemorrhagic venous infarcts due to venous thrombosis of superior sagittal sinus or great vein of Galen |
| Hemorrhagic origin |
Subdural hematoma |
| Subarachnoid hemorrhage |
| Intracerebral hemorrhage |
| Multiple microbleeds (especially those subcortical ) |
| Focal Disease due to Strategic Lesions |
Small infarcts located in functionally important regions such as: |
Mesial temporal |
| Caudal and thalamic infarcts Fronto-cingular infarcts |
| Angular gyrus infarcts |
| Strategic areas of white matter |
Table 1: Morphological lesions causing vascular cognitive impairment [11].
Cerebral microbleeds are easily identified with gradient echo or Swan MRI sequences; these are histopathologically defined as extravasation of blood into the perivascular space. When located in lobar regions, they are related to cerebral amyloid angiopathy, with accumulation of amyloid protein in the wall of arterioles, which become weak and prone to rupture. Occasionally, usually in the context of a genetic alteration, a patient may have hundreds of microbleeds and their presence is a predictor of cognitive impairment [39].
Recommendations
It is critical to establish the size and location of vascular injury in all patients with CVD. It is also necessary to determine the type of vessel affected (large or small), since the damage of the latter includes lacunar infarcts, microbleeds, hyper-intensities or perivascular spaces, which have an impact on the prognosis and treatment of cognitive disorder.
Risk Factors of Vascular Cognitive Impairment
Demographic and genetic factors
The most important demographic predictor for VCI is age; thus, the aging of Latin American populations probably will lead to a higher incidence of VCI. There is no an apparent difference by sex or ethnic factors, and there is a need for reliable geographical studies in Latin America [40-42].
Regarding genetic factors, the role of ApoE epsilon-4 has been studied for its relevance in Alzheimer’s disease, but no association with VCI has been established [43,44]. Among genetic diseases, CADASIL stands out, being a non-amyloid small vessels disease associated with cognitive impairment and evidence of alteration in NOTCH gene [45,46].
Lifestyle factors
Diet: It has been proposed that the intake of antioxidants as those contained in vitamin E, vitamin C and beta carotenes in diets including fruits and vegetables, or as supplements, may reduce the risk of VCI [47-49]. However, controlled clinical trials do not show benefit of consuming these antioxidants in the preservation of cognitive function or its impairment reduction [50-52]. The N-3 polyunsaturated fatty acids in fish have antioxidant and antiinflammatory properties, and they are also the major components of phospholipids of brain cell membranes. Investigations in this respect have shown that the intake of fish for several years (3-6 years) is associated with lesser decline in cognitive function [53- 59]. Therefore, a diet rich in polyunsaturated fatty acids in fish oil is recommended. The so-called Mediterranean diet has shown increasing evidence that may reduce cognitive impairment, and it is reasonable to recommend it [60-63].
Obesity: Body mass index (BMI) is directly associated with VCI. The Framingham Offspring Study showed that the higher the waist-hip ratio was the lower cognitive function during a 12- year follow-up. It is reasonable to recommend the body weight control for preserving cognitive function [64-68]. An increase of BMI is associated with obstructive sleep apnea (OSA), a factor of vascular risk, and a frequent cause of cognitive function impairment. OSA causes subcortical lesions in withe matter by lesions in small brain vessels [69].
Exercise: Regular, long-term physical activity, including vigorous exercise and walking, has been associated with better cognitive function and less impairment with age. Although the LIFE study did not yield predicted results for moderate physical activity and cognition improvement in sedentary adults aged 70-89 years, it did show that those at 80 years and older experienced benefits in executive function [70]. Physicians should encourage patients of all ages to optimize their levels of physical activity for the entire life to reduce the risk of developing conditions affecting cognition and other diseases [71]. Therefore, there is a favorable evidence to recommend routine physical activity as a preventive strategy in VCI [72-82].
Smoking: This is a well-known risk factor for ischemic and hemorrhagic cerebrovascular events. Several prospective studies have confirmed higher risk of cognitive impairment in smokers, particularly for some cognitive domains. The evidence indicates that smoking discontinuation is essential for individuals at risk of VCI [83-88].
Alcohol: Some investigations have shown certain benefit in cognition for alcohol users, especially in low to moderate amount, compared to those who do not drink or do it occasionally. For some authors, the benefit would occur primarily in older subjects. However, there is no conclusive data on the amount of alcohol, gender or cognitive domains impacted. Alcohol abuse is associated with all types of CVD. Although moderate alcohol consumption is recommended in patients with VCI [89-95], the experience shows that it is impossible to limit alcohol consumption in patients with cognitive deficit with amnesic predominance; thus, alcohol total abstinence is advisable in these patients.
Depression
Depression may be a comorbidity, a prodromal factor or a consequence of VCI rather that a factor leading to cognitive impairment [96-99]. The Three-City study did not demonstrate an association between VCI and major depression during a four-year follow-up [100]. The Cardiovascular Health Study also could not confirm it [101]. Therefore, depression is not currently considered as a risk factor for VCI. Late-life depression (defined as that beginning after age 60) in a cross-sectional study showed that individuals with depression were more likely (20%) to develop vascular dementia [102].
Chronic inflammation
Inflammation is a cardinal process associated with many risk factors of neuronal and vascular damage. Plasma levels of inflammatory proteins, specifically alpha-anti-chymotrypsin and C-reactive protein (CRP), increased prior to the development of vascular dementia after 8 years of follow-up; the CRP levels are high up to 25 years before the onset of vascular dementia. In another study with 4-year follow-up, it was established that a combination of high CRP and interleukin-6 levels resulted in a 3-fold increased risk of developing vascular dementia [103-105].
Chronic renal failure
Several studies with different populations suggest that patients with moderate and severe chronic renal disease have an increased prevalence of cognitive impairment affecting multiple domains. Chronic renal failure is associated with hypertensive encephalopathy and increased risk of CVD. A close relationship between the reduction of glomerular filtration rate and the presence of VCI has been demonstrated. In the Cardiovascular Health Study [106], moderate chronic renal disease was associated with a higher incidence of vascular dementia. However, the association between chronic renal disease and cognitive impairment may be confounded because they share vascular risk factors for small-vessel disease [107-109].
Systemic arterial hypertension
Hypertension is the main risk factor for ischemic and hemorrhagic CVD. The relationship between blood pressure and cardiovascular risk is consistent and independent of other risk factors. More than two thirds of people over 65 are known to be hypertensive. Appropriate control of blood pressure contributes not only to prevent CVD but also to reduce cardiac and renal damage. Antihypertensive treatment has been associated with a reduction in the incidence of CVD from 44% to 35%. Systolic hypertension is recognized as a risk factor to develop VCI and dementia, not only of vascular type but also degenerative dementia such as Alzheimer disease. Hypertension exposes the cerebral microvasculature to pulsatile flow and pressure leading to endothelial damage which causes lipohyalinosis and fibrinoid necrosis. The resulting disruption of perfusion leads to the development of lacunar infarcts or chronic ischemia expressed as a subcortical vascular lesion (leukoaraiosis). There are several cross-sectional and cohort studies demonstrating the close relationship between systolic or diastolic arterial hypertension in the person’s half-life and the development of vascular cognitive impairment [110-112].
Studies have shown that there is a “J” or “U” relationship, where the effect of controlling hypertension to prevent the development of VCI is effective when this is done from the middle of the life, but not when it is treated late at an advanced age.
Diabetes mellitus
People with diabetes mellitus (DM) have an increased risk of developing CVD, with a relative risk between 1.8 and 6 depending on the type and severity of DM. Also, patients with DM are about twice as likely to develop dementia. Subjects with DM have lower scores in tests of memory, processing speed and executive functions; these are more apparent with poor glycemic control. MRI studies have shown that patients with DM have more lacunar infarcts and hippocampal atrophy. DM is likely to influence cognitive impairment, independently of its role as a vascular risk factor. Hyperglycemia acts through advanced glycation terminal products that have been found in neuritic plaques and neurofibrillary tangles in the early stages of AD. The Rotterdam study indicates that DM doubles the risk of AD and VCI, particularly in subjects with APOE-4 genotype. The presence of repeated and intermittent hypoglycemic episodes in insulintreated patient causes hippocampal atrophy and cognitive deficit [113-115].
Dyslipidemia
The role of lipid disorders on VCI remains controversial. Dyslipidemias are likely to participate in VCI indirectly through the development of atherosclerosis. Autopsy studies have revealed that patients with AD show atherosclerosis in the circle of Willis in 77% of cases. However, most of the studies assessing the effectiveness of statins in patients with dementia show no improvement [116,117].
Subclinical atherosclerosis
Subclinical atherosclerosis is noninvasively detected, primarily by high-resolution ultrasound measurement of carotid intima-media thickening (IMT). It may also be assessed through studies that measure arterial stiffness (essentially the carotid-femoral pulse wave velocity). IMT has been related with most of the vascular risk factors and the development of vascular events (CVD, coronary). A significant inverse relationship between IMT and cognitive function has been observed; the greater the IMT, the lower the cognitive capacity regardless of age, sex and traditional vascular risk factors. Likewise, studies using carotid pulse wave velocity have found greater arterial stiffness in subjects with cognitive impairment [118,119].
Cardiovascular disease
VCI risk has been associated with atrial fibrillation (AF), heart failure, chronic renal disease and coronary and peripheral arterial disease. AF impacts on the development of VCI not only through the production of cardioembolic ischemic cerebral events, but also with an increased risk of AD and VCI, independently, although the mechanism is unknown. It has been shown that cerebral volume in subjects with AF is lower than in controls without AF. It has been stated that this is a consequence of hypoperfusion when AF has a rapid ventricular response or because of the presence of microembolism. One of the causal factors of AF is unsuspected and untreated OSA. On the other hand, the reduction of cardiac output has been associated with a decrease in cognitive functions, in particular executive functions. It has been proposed that chronic hypoperfusion is the major contributor but more studies should be conducted. Coronary artery disease is also associated with an increased risk of VCI [120,121]. In addition, coronary artery bypass surgery is associated with early cognitive impairment (post-surgery) and increased risk of dementia in the median term. Finally, peripheral arterial disease measured through the ankle-arm index is also associated with a higher risk of vascular dementia [122,123].
Sleep disorders
From Nurses’ Health Study [124], it was observed that women who sleep 5 hours or less or those who sleep 9 hours or more at night had a higher risk of MCI compared to women who sleep 7 hours. OSA is one of the risk factors most frequently missed when evaluating patients with VCI [125].
Hyperhomocysteinemia
Several studies have shown that high levels of homocysteine are associated with an increased risk of cardiovascular events, either coronary or cerebrovascular. There is also evidence that hyper- homocysteinemia is related to VCI, either through cerebrovascular damage or directly. Hyperhomocysteinemia may be determined by genetic-environmental factors, mutations in critical metabolism enzymes, mainly metylentetrahydrofolatoreductase (MTHFR) and deficiency or low levels of vitamins involved in metabolism such as vitamin B12 and folic acid. Some families with MTHFR gene mutations may have a history of depression, suicide and alcoholism, as well as elevated homocysteine. The treatment of hyperhomocysteinemia with cobalamin (vitamin B12), pyridoxine (vitamin B6) and folic acid (vitamin B9) in patients with minimal cognitive deficit stops the progression to dementia and cerebral atrophy typical of AD [126-128].
Recommendations
VCI has been associated with several preventable vascular risk factors such as hypertension, sedentary lifestyle, obesity, smoking, diabetes mellitus, hypercholesterolemia and obstructive sleep apnea. Management of these risk factors remains a practical approach to reducing VCI.
Diagnostic Methods for Vascular Cognitive Impairment
Diagnostic neuropsychological tests in vascular cognitive impairment
Cognitive impairment is characterized by a wide range of cognitive deficits, but there is a predominance of executive dysfunction [129,130]. Therefore, the neuropsychological protocols should be sensitive to various domains, with an emphasis on executive function assessment. For Latin American population, it is important to consider several aspects to conduct the cognitive assessment: (a) education level; (b) standardized and validated tests for study population; (c) time to conduct it, and (d) personnel who will conduct assessments (doctor, psychologist).
There are no specific and universally accepted tests for VCI diagnosis. There are three recommended protocols lasting 60 minutes, 30 and 10 minutes. The 60-minute test may be used in studies requiring a detailed analysis of capabilities in the cognitive domains. Physicians may use the following tests during visits:
30-minute tests: The clinical screening tools in patients with suspected VCI include the following [131]:
1. Phonological fluency
2. Semantic fluency
3. Montreal Cognitive Assessment (MoCA)
4. Digits in regression
5. Mini-Mental State Examination (MMSE)
6. Trial Making Test A and B (TMT A and B)
7. Hopkins Verbal Learning Test (HVLT-R)
10-minute tests: These are instruments to be used by primary care physicians, nurses, and other healthcare professionals, to allow rapid detection of VCI:
1. Mini-Mental State Examination (MMSE): This is a widely used method to estimate the intellectual status but insufficient to assess the executive function; its 3-word memory test is insensitive to early detection of memory impairment in patients with VCI.
2. MoCA (Montreal Cognitive Assessment): This short test designed to detect cognitive impairment in older adults has a maximum score of 30 points. The MoCA test is sensitive to subcortical damage. It may also be administered via telephone [132,133]. The MoCA in Spanish (MoCA-S) has been validated in some countries of Latin America. In Mexico, validation showed a sensitivity of 80% and specificity of 75%, with cut-off point of 26 for MCI (area under the curve 0.886 p <.001). For the dementia group, it showed a sensitivity of 98% and specificity of 93%, with cut-off point of 24. (Area under the curve 0.998 p<0.001) [134]. In Colombian elderly subjects with low education, the MoCA-S had a high reliability but scores were strongly dependent on years of education, social and cultural factors [135]. A compensation of 3-4 points was needed for subjects with<6 years of education [136].
Neuropsychological tests proposed for VCI, by cognitive domain
Execution/activation (planning, decision making, working memory, response to feedback or error corrections, inhibition/ predominant habits, mental flexibility):
1. Semantic fluency [137]: A test related to left posterior parietal-temporal region.
2. Phonological fluency [138]: Reflecting integrity of left dorsolateral frontal region.
3. Wechsler Adult Intelligence Scale (WAIS) – Digits: It evaluates processing speed and activation.
4. Trail Making: It is a test of exploration, visual-motor tracking, divided attention and cognitive flexibility. The test is very sensitive to the presence of cognitive impairment [139].
5. Hopkins Verbal Learning Test (HVLT-R) [140]: It may provide strategic learning measures reflecting the dorsolateral frontal function in addition to episodic memory rates.
6. Frontal Assessment Battery (FAB): It includes six subtests, which correlate with frontal lobe function. The performance in these six subtests may give a composite global score, which assesses the severity of the dysexecutive syndrome [141].
7. Clock drawing test [142,143]: The common errors in executive function showed in this test include incorrect placement of the hands, graphic problems (diffuse lines, small circle, correction attempts) and difficulties in spacing numbers.
Visuospatial (visoconstructive skills, visual perception): This neuropsychological test is performed through Rey-Osterreith complex figure [144]. This task requires organization and visuoperceptual skill. The primary visuospatial test is administered by copying the Rey-Osterreith figure, and the memory test was selected as a supplementary measure.
Language: Evaluation includes expressive language such as naming objects, finding words, fluency, grammar and syntax, as well as receptive language. Common language tests are the following:
1. The Boston Naming Test (BNT) [145]: It diagnoses the presence and type of aphasic deficit, allowing global assessment of difficulties in all the areas of language. Evaluation of naming capability is by visual confrontation.
2. Semantic fluency: It may serve as a less structured lexical retrieval task.
Learning and memory: It includes immediate memory, recent memory, free recall, evoked recall and recognition, as well as long-term semantic and autobiographical memory and implicit learning. Tests include:
1. HVLT-R (Hopkins Verbal Learning Test – Revised): This is the preferred learning test. Its strengths include a multiple-choice form and a short administration time. The HVLT-R does not include an interference list or a key recall. Clinical studies have shown that it is sensitive to cognitive impairment related to VCI.
2. Wechsler Memory Scale: It allows assessment of verbal learning capability, short- and long-term retention and recognition capability, as well as other more specific aspects such as the interference effect or the trend to respond with false positives; it is sensitive to VCI [146,147].
3. RAVLT (Rey Auditory Verbal Learning Test) or Rey test for verbal and auditory learning: It assesses recognition, interference, learning curve, immediate and delayed memory.
To establish a difference between static cognitive deficits following a cerebrovascular disease and the progressive dementia syndrome, it is advisable to conduct serial neuropsychological tests, usually after a year of evolution.
1. California Verbal Learning Test-II: Useful for evaluating episodic verbal learning and memory [148].
Recommendations
There are no specific and universally accepted tests for the diagnosis of VCI. There is no ideal neuropsychological test and the assessment should include sensitive tests for each type of cognitive disorder. MoCA is a sensitive screening test to detect VCI and should be used to detect early forms of this disorder. Other recommended tests include those of semantic and phonological fluency, FAB and clock test. All the tests should be standardized and validated for the study population. The assessment of illiterate or low education patients is a problem that requires the urgent development of appropriate evaluation methods in Latin America.
Neuroimaging studies
The main neuroimaging choices to study VCI are MRI and CT scan. MRI is particularly sensitive in the evaluation of cognitive disorders characterized by small-vessel lesions. The minimum acceptable field strength is 1.0 Tesla, but ≥ 1.5 T is preferred. The following sequences are required: weighted in T1 and T2, FLAIR, and gradient echo. The first 3 sequences provide information on the anatomy, the presence of an infarct and other pathologies, while the gradient echo identifies the presence of bleeding, including small, chronic and/or acute hemorrhages. Neuroimaging findings should be interpreted in a clinical context considering the infarct nature, severity and location. The use of Arterial Spin Labeling (ASL) imaging for cerebral blood flow in non-contrast MRI is a promising technique in VCI.
Neuroimaging-supported criteria to determine vascular dementia: Although it is accepted that VCI may rarely occur without evidence of cerebral infarcts in the neuroimaging studies, these lesions are usually present. A single strategic stroke or an extensive stroke having effects in several cognitive domains may be necessary to cause a VCI. If multiple-stroke disease is a cause of VCI, at least one should occur outside the cerebellum. Ideally, there should be a temporary association between the stroke and the onset of cognitive impairment, in such a way that the ischemic lesion precedes the impairment within previous three months, and that this impairment extends beyond this period.
California criteria [26]: Two or more ischemic infarcts, with at least one of them outside the cerebellum, are required to diagnose vascular dementia.
NINDS-AIREN criteria [25]: Multiple large vessel infarcts or a strategic infarct (occurred at angular gyrus, thalamus, basal forebrain, or in the posterior or anterior carotid region) or lacunar infarcts in the white matter or basal ganglia, or extensive periventricular lesions in the white matter are required.
Newcastle criteria [35]: The presence of >3 lacunar infarcts should be appropriate evidence of causality, especially with coexisting white matter disease.
Characteristics of lacunar infarcts in neuroimaging [149]: Lacunar infarct is better detected with a FLAIR sequence in MRI, which shows the lesion as a small hypointense area surrounded by a hyperintense halo, although the fluid in the central cavity is not suppressed in FLAIR sometimes and may appear completely hyperintense. Lacunar infarct is commonly considered as a 3-15 mm lesion, but definitions vary with maximum diameters of 2 cm. The VCI harmonization group recommends consider “lacunes” up to 1 cm, with the main characteristic of being ischemic lesions or small hemorrhages in the deep white matter of a perforating arteriole. Given their small size, the MRI sequence with adjacent slices ≤ 4 mm is required to detect adequately these lesions. In CT, lacunar infarcts are small hypodense lesions (i.e., well defined), but given the poor CT spatial resolution, these lacunar lesions may remain unnoticed. It is important not confuse lacunar lesions with Virchow-Robin perivascular spaces, although these findings may really represent and early stage of small vessel disease with an underlying microvascular degeneration.
Vascular lesions in sub-cortical white matter: In CT, white matter lesions are hypodensities (leukoaraiosis). In MRI, they are hypointense areas in T1 and hyperintense in T2-weighted images. Subcortical vascular leukoencephalopathy may be focal or multifocal, and as these types of lesions become more extensive, they may converge and involve a large area of white matter. Basal ganglia and thalamus also show these lesions. When vascular leukoencephalopathy is mild, these lesions appear as small “caps” in frontal or occipital horns of lateral ventricles, or as thin “rings” surrounding these structures [150]. However, these images are not specific to vascular leukoencephalopathy, as comparable patterns may be observed in several pathologies, including multiple sclerosis, cerebral edema, neurosarcoidosis, brain lesion caused by radiation, etc. Despite the diverse listing of differential diagnoses, there is clinical evidence and pathological data suggesting that most of these brain lesions in older subjects have an ischemic origin. These lesions are caused by arteriolosclerosis, lipohyalinosis and fibrinoid necrosis of the small vessels, and particularly along the perforating arteries, with or without occlusion. Vascular leukoencephalopathy is highly prevalent in the brains of older people and even middle-aged individuals; thus, only extensive lesions are clinically significant. If vascular leukoencephalopathy is focal and visible only in T2, it is unlikely to be significant enough to explain the development of cognitive disorder. Some researchers have suggested that at least 25% of total white matter should be affected to support a vascular dementia diagnosis. Therefore, it is difficult to have precise rules for relating leukoencephalopathy to mild VCI or vascular dementia. A general rule is that these lesions should be extensive and confluent and the above descriptions are considered as patterns of subcortical vascular involvement. With the development of new MRI techniques such as diffusion tensor imaging, it has been shown that white matter that appears normal in T2 may also have anisotropy or abnormal diffusivity matching with the neuropathology and be relevant to support the presence of cerebrovascular damage in the presence of cognitive alterations [151,152].
Intracranial hemorrhagic lesions: Cognitive disorders have been associated with intracerebral hemorrhage, subarachnoid hemorrhage, and subdural hematoma. VCI has also been associated with cortical and subcortical microbleeds, which may be related to hypertension or cerebral amyloid angiopathy.
This angiopathy is a consequence of the accumulation of Aβ40 (different from A42 that forms the plaques in AD), which is deposited in the intima and the media of the vessel, producing fragility and hemorrhage, but also predisposes to thickening of the artery, which causes ischemia [153]. These lesions are better visualized in MRI Eco-GRE sequence. Microbleeds associated with hypertension are in the deep brain nuclei and brainstem, while those associated with amyloid angiopathy and Alzheimer’s disease has usually a lobar location. As microbleeds are not uncommon in cognitively normal older adults, the attribution of VCI to microbleeds should be done only when these lesions are numerous and after careful exclusion of other causes of cognitive impairment.
Cerebrovascular assessment by other studies: Doppler ultrasonography technology provides useful information on the state of cervical and intracranial arteries, complementing the information of parenchymal brain lesion identify by neuroimaging studies. Transcranial Doppler is a dynamic study of cerebral blood flow velocities that helps to determine functional characteristics and to deduce structural aspects as well. Brain perfusion is also assessed through single photon emission computed tomography and CT with xenon contrast. Positron emission tomography (PET) allows imaging through regional glucose metabolic rates, using 18F-fluorodeoxyglucose (FDG-PET). It is useful for establishing the differential diagnosis of some types of cognitive disorders. There is no characteristic pattern in patients with vascular dementia, and its current usefulness is to rule out another type of dementia [154]. More recently, amyloid imaging with compounds such as radiolabeled Pittsburgh Compound B (C-PIB) has gained a great interest and has been proposed as a biomarker for AD imaging. ASL imaging in brain MRI measures cerebral blood flow in a noninvasive way, it does not require any contrast injection, and does not expose the patient to radiation; furthermore, there is a good correlation between ASL and FDG-PET.
Recommendations
It is essential to have a neuroimaging study when assessing the patient with cognitive impairment for probable vascular etiology. Brain CT should be performed in all cases. Cerebral MRI is the ideal study to characterize small-vessel lesions and it should be requested considering the individual clinical context for each patient.
Genetic and inflammatory biomarkers and their neuropathological correlation in vascular cognitive impairment
In patients with VCI, several biomarkers have been investigated for early detection, to discriminate neuropathological findings, to estimate prognosis and to monitor disease progression or therapeutic response [155,156]. In these circumstances, markers related to genetic factors and inflammatory mediators involved in the etiopathogenesis of VCI are of interest.
The analysis of cerebrospinal fluid (CSF) biomarkers, along with clinical and neuroimaging information, may enhance the diagnosis of different brain disorders causing cognitive impairment [156]. Vascular dementia is generally related to the following biomarkers in CSF:
Total tau: It is a dynamic marker of the intensity of axonal degeneration/damage. Its levels increase in Creutzfeld-Jakob disease, and it may also be found in dementia associated with VCI, cerebral trauma and AD [157-159]. The usefulness of tau protein in the diagnosis of vascular dementia is to exclude other etiologies, so that there are no cut-off points for the diagnosis of vascular pathology [160].
Light sub-unit of neurofilament protein: It is the best biomarker in CSF for subcortical axonal damage/degeneration. It occurs at high concentrations in vascular dementia, frontotemporal dementia and different inflammatory disorders (multiple sclerosis, dementia- AIDS complex). The combination with an AD biomarker indicates the presence of mixed dementia [155,161-164].
CSF and serum albumin concentration index: It is the most defined biomarker of integrity of the blood-brain barrier. There is usually an elevation of this ratio in patients with vascular dementia (particularly due to subcortical vessel disease) [156,159,164].
Levels of tumor necrosis factor alpha (TNF-α): This proinflammatory cytokine mediates myelin damage. Its levels increase in patients with subcortical vascular dementia and correlate to sulfatide levels (a white matter degradation marker). It is found potentially high in subcortical vascular dementia [165].
Sulfatides: They identify possible white matter demyelination.
Metaloproteases [166]: They increase when there is vascular or white matter damage. It has been demonstrated that high levels of some of them allow prediction of the evolution of vascular impairment. High levels of metalloproteinase MMP-9 are associated with worse prognosis for VCI compared to those with a stable disease and low MMP-9 levels. According to imaging studies, there is also a positive correlation between MMP-9 mean values and the volume of infarct or ischemia. MMP-9 and MMP-2 levels depend on the occlusion time of vascular bed, which may extend, expand or cause worse vascular damage.
Other cytokines: The assessment of interleukin IL-1 is considered important, along with tumor necrosis factor alpha. It may be associated with worsening of vascular impairment. They could also be correlated to the measurement of IL-10 and IL-1ra (IL-1 receptor antagonist interleukin) by exerting a potential neuroprotective effect. IL-6 is present in acute ischemia and reflects the extent of the infarct; it could also be correlated to severity of vascular damage in the patient with VCI.
Anti-inflammatory prostaglandin 15-Dpgj2: The high levels of this prostaglandin may correlate with better prognosis in the evolution of VCI.
Homocysteine: Its persistent elevation matches with the potential presence of a deleterious inflammatory process that may perpetuate and enhance the vascular damage with the corresponding progression of cognitive impairment [167].
C-reactive protein: It is known to be associated with a poor long-term functional prognosis when it is increased in the acute phase of the cerebrovascular event. Although this marker cannot predict the stroke progression, it is an excellent, independent predictor of mortality and morbidity in CVD evolving into VCI.
Non-specific biomarkers used for distinctive diagnosis of vascular cognitive impairment: The finding in CSF of low levels of amyloid beta 42 amino acids, with high levels of hyperphosphorylated tau, or biochemical signs of neuro-inflammation (increased white blood cells, production of IgG or IgM) indicates nonvascular dementia and may be useful as negative biomarkers for the diagnosis of pure vascular dementia. However, the implementation of CSF biomarkers in the diagnostic process of vascular dementia requires standard methods in the collection, storage and measurement of the sample [155-157].
Recommendations
At present, there is no biomarker capable of establishing the diagnosis of VCI. The utility of these biomarkers is to confirm the presence of neuronal damage or inflammation. More studies are needed to identify a sensitive and specific marker to establish the early diagnosis of VCI.
Mixed Dementia
The concept of mixed dementia refers to the coexistence of a typical degenerative disease (e.g., Lewy body dementia, frontotemporal dementia, Alzheimer’s disease) and vascular dementia. Alzheimer’s disease shows the strongest evidence of its interrelation with vascular lesions.
The term mixed dementia represents a challenge for clinicians and even for neuropathologists, and expresses the increasing difficulty to isolate a “pure” type of dementia (vascular versus Alzheimer’s), especially in preclinical stages. At the same time, it represents a model for the study of twoway interaction between vascular lesions of different types (atherosclerosis, arteriolosclerosis, amyloid angiopathy) and neuronal degeneration [168]. Vascular lesions can cause both neurodegeneration and its typical markers (e.g., neurofibrillary tangles, β-amyloid), as similarly neurodegenerative pathology can accumulate β-amyloid compromising the cerebral vasculature and cause vascular lesions. As long as the classification of pure neurodegenerative or vascular forms remains as the initial step to classify dementias, we will be forced to define a “hybrid” called “mixed dementia”. The proposed approach for the classification is described in Figure 2 and consists of determining first whether dementia (meeting the accepted criteria) is associated or not with overt cerebrovascular lesions, to rule out pure forms in this way. Subsequently, a distinction is established among those showing evidence of vascular lesion, those having more clinical and paraclinical features of degenerative dementia despite vascular lesions, and those having more clinical and paraclinical characteristics of vascular dementia but also with evidence of neurodegenerative pathology.
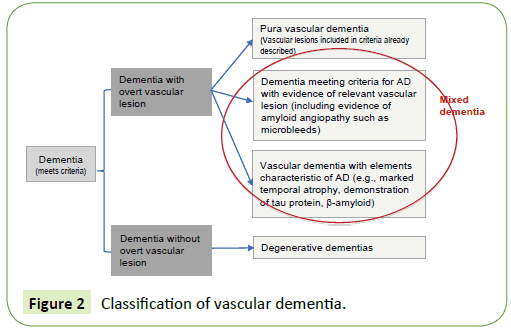
Figure 2: Classification of vascular dementia.
Association between cerebrovascular disease and Alzheimer’s disease
Epidemiological studies show that Alzheimer’s disease and cerebrovascular disease share similar risk factors including hypertension, diabetes, smoking, apolipoprotein E epsilon 4 isoforms, hypercholesterolemia, chronic nephropathy and, mainly, age [169,170]. Cardiac risk factors, in particular atrial fibrillation and congestive heart failure, have also been associated with the pathogenesis and progression of Alzheimer’s disease [168-177].
A study in Mexican patients with Alzheimer's disease and mixed dementia demonstrated after multivariate analysis that hypertension (OR: 1.92, p= 0.009), white matter disease (OR: 3.61, p= 0.001) and lacunar infarcts (OR: 3.35, p = 0.014), were associated with mixed dementia [178].
Different studies have documented the overlapping of cerebrovascular disease and Alzheimer’s disease, an association even stronger in Braak stages with a lower density of neurofibrillary tangles. The autopsy study with 5,715 cases of the National Alzheimer’s Coordinating Center confirmed data on the prevalence of cerebrovascular disease and Alzheimer’s disease, and the deleterious additive or interacting effect of vascular pathology and Alzheimer’s disease on cognition [179-181].
Likewise, aging itself has a deleterious effect on the cerebral vasculature similar to that caused by Alzheimer’s disease, as when drainage of soluble β-amyloid is affected it leads to accumulation in vascular walls and parenchyma, with the already known consequences on homeostasis of neuronal environment and cerebral perfusion [106,182]. In addition, the presence of cerebrovascular disease has been associated with worse cognitive performance in patients with AD, and neuropathological studies have documented that cerebrovascular disease reduces the dementia threshold in subjects with pathological diagnosis of AD [183,184]. Thus, it has been proposed that CVD contributes to neuropathological changes of AD, including selective cerebral atrophy and accumulation of abnormal proteins such as β-amyloid. On the other hand, subcortical AD and CVD may independently affect cortical atrophy [185-187].
Type of vascular lesions more associated with mixed dementia
In brains with AD and mild CVD (primarily degenerative dementia), most of the cerebrovascular lesions are lacunar infarcts in basal ganglia and white matter, with multiple microinfarcts. This pattern of topographic distribution of cerebrovascular lesions is very similar to that observed in “pure” vascular dementia (vascular dementia without histopathology of AD greater than expected for the age). Sixty-eight percent are lacunar infarcts involving thalamus or hippocampus, and only 32% are extensive cortico-subcortical infarcts.
In contrast, mixed dementia may be accompanied by lobar infarcts and multiple cortico-subcortical lesions (57%) rather than subcortical lacunar infarcts or strategic infarcts (43%). This suggests different pathogenic mechanisms between these two entities, which give greater importance to microangiopathy in pure vascular dementia or AD with mild cerebral vascular disease than in mixed dementia, as described in Table 2, which compares common lesions in AD, VD and mixed dementia [188].
| Pathology |
AD (%) |
VD (%) |
Mixed dementia (%) |
| Total infarcts |
10-20 |
100 |
30-40 |
| Small vessel disease |
Approx. 50 |
>50 |
>50 |
| Lacunar infarcts |
30-46 |
70 |
60-70 |
| White matter pathology |
40 |
80 |
70-80 |
| Intracerebral hemorrhage |
10-15 |
15 |
10 |
| Cerebral amyloid angiopathy |
98 |
30 |
Approx. 90 |
| Loss of cholinergic markers |
75 |
40 |
Approx. 70 |
| Atherosclerosis |
45-60 |
60 |
Approx. 60 |
AD: Alzheimer’s Disease; VD: Vascular Dementia.
Adapted from Jellinger KA, Attems J (J Neurol Sci 2007, 257: 80-87).
Table 2: Vascular lesions comparison between AD, VD and mixed dementia.
The combination of two or more pathological processes may influence the severity of cognitive impairment in such a way that cerebrovascular lesions may unmask dementia at preclinical stage in patients with Alzheimer’s disease. However, apparently small cerebrovascular lesions occurring in 10-50% of the adult population without cognitive impairment cannot cause dementia by themselves.
Physiopathogenesis proposed for mixed dementia
Microvascular changes in the brain of older people and AD induce cerebral perfusion alteration, in particular, a decrease in regional blood flow, reduction of glucose transport and utilization, with loss of vascular innervation and a special impact on the deficits in cholinergic neurotransmission in the case of AD. Likewise, there is damage in neurovascular regulation, ultrastructural changes in capillaries and basement membranes due to deposition of β-amyloid, with disruption of the blood-brain barrier and alteration of β-amyloid clearance. The succession of these events and other deleterious effects create a vicious circle, which finally produces structural disintegration through infarcts, lacunar lesions and vascular lesions in white matter, which compromise neuronal metabolism, with mitochondrial energy deficit and oxidative stress, promoting the degradation of proteins, lesions in cytoskeleton with formation of neuritic plaques and deposition of β-amyloid. These factors induce cerebral atrophy and the consequent cognitive impairment.
On the other hand, atherosclerosis and amyloid angiopathy cause changes in the self-regulation of microvasculature, leading to loss of myelin, frequently observed in elderly people’s brains, suggesting shared risk factors for all pathological changes observed in AD and cerebrovascular disease. White matter lesions can be caused by both cerebral vascular disease (hypoperfusion) and AD (retrograde degeneration). These lesions progress with age, represent a considerable risk factor for cognitive impairment and lead to impairment in frontal functions regardless of their location [189-191].
The most common clinical expression in neuropsychological tests is combined; i.e., they exhibit characteristics of a subcortical cognitive profile, as well as of cortical. However, regardless of the predominance of the clinical pattern showed by the patient, in almost all the cases there is a clear compromise of frontal and prefrontal functions.
Diagnostic criteria for mixed dementia
The clinical criteria for the diagnosis of mixed dementia are diverse and pose difficulties for the study of this condition as an independent entity. The International Classification of Diseases (ICD-10) does not consider mixed dementia an isolated entity but includes it in the AD section “G30.8: Other Alzheimer’s-type Diseases (atypical and mixed forms)”.
The classification by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) [192] does not consider mixed dementia in its classification, while the criteria of the Alzheimer’s Disease Diagnostic and Treatment Centers (ADDTC) require the evidence of a CVD closely related to dementia (referred to without a time framework), in addition to the typical pathology of AD [26]. The NINDS-AIREN criteria suggest to demonstrate an evidence of memory compromise and at least two other cognitive areas, in addition to an evidence of cerebrovascular lesion (focal neurological signs and infarcts/lesion in brain white matter imaging), and that the onset of dementia occurs within 3 months from the occurrence of the cerebrovascular event [25].
In 2011, the AHA/ASA recognized that vascular and degenerative pathologies are frequent and coexisting disorders in elderly people, and that separately each adds to the other the possibility of developing cognitive impairment and dementia with overlapping clinical and neuroimaging phenotypes. Additionally, there is a need to resolve the concerns by establishing a relationship between neuroimaging findings and postmortem studies [169]. First, by improving the resolution capacity of neuroimaging studies, as they currently can detect macroscopic infarcts of more than 3 mm, but fail to detect microscopic infarcts and small vessels disease (such as arteriolosclerosis). Second, some vascular pathologies may represent vascular or degenerative process, as degeneration of white matter, measured in FLAIR sequence and by diffusion, as well as microbleeds, measured by echo-gradient sequence, are associated with both cognitive vascular disorder and AD [193-195], and pathology studies have shown that white matter degeneration and microbleeds are related to lipohyalinosis [196,197].
On the other hand, changes in hippocampal volume may be related to AD-type or vascular pathology [198] in such a way that the atrophy of hippocampus may occur in response to a degenerative or vascular process [199]. The different criteria used so far for mixed dementia around the world are detailed comparatively in Table 3 [24-26,200-202].
| Source |
Criteria |
| Hachinski scale |
Scale based on clinical data: £ 4 = AD, = 7 = VD; middle score of 5 or 6 = MD |
| International Classification of Diseases (ICD 10th edition) |
Cases metting criteria for AD and VD. |
| Diagnostic and Statistical Manual of Mental Disorders (DSM-V) |
Cases with criteria for Alzheimer-type primary degenerative dementia and clinical or neuroimaging data for VD. |
| Alzheimer’s Disease Diagnostic and Treatment Centers (ADDTC) |
Presence of isquemic cerebrovascular disease and a second cerebral or systemic disorder. |
| National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherche´ et l’Enseignement en Neurosciences (NINDS-AIREN)* |
Typical AD data associated with clinical and radiological evidence of cerebrovascular disease. |
AD: Alzheimer’s Disease; VD: Vascular Dementia. MD: Mixed Dementia.
*They do not call it MD but AD with cerebrovascular disease.
Table 3: Criteria for mixed dementia.
Gold et al. [203] compared the clinical findings to the neuropathological diagnosis in 113 elderly subjects with dementia undergoing autopsy. The subjects were neuropathologically classified with mixed dementia if they met the criteria for AD and for vascular dementia. All the clinical criteria examined had a different behavior when diagnosing mixed dementia behavior. The proportion of cases neuropathologically diagnosed with mixed dementia which have been clinically classified as vascular dementia was 54% for ADDTC, 29% for the NINDS-AIREN criteria, and 18% for the Hachinski scale. The Hachinski scale allowed to exclude more cases of mixed dementia but failed to identify many cases of vascular dementia. The ADDTC and NINDS-AIREN criteria were more sensitive to detect vascular dementia, but less able to distinguish between vascular dementia and mixed dementia. Mixed dementia was better excluded with NINDS-AIREN than with ADDTC. The authors reported that mixed dementia has a significant effect on the acuity of the diverse clinical criteria. Recent data from the same group have confirmed that clinical criteria for vascular dementia are not equivalent to those for mixed fementia and work very differently with respect to mixed dementia detection. Furthermore, restrictive criteria such as ICD -10 or the probable category of ADDTC and NINDS-AIREN do not correlate significantly to histopatological diagnoses. In a metaanalysis, Moroney et al. found that Hachinski scale distinguished well between AD and VD, but it had difficulties with mixed dementia diagnosis [204].
From the above mentioned, we conclude that there are no validated and accepted clinical-pathological criteria for the diagnosis of mixed dementia and therefore there are only recommendations for its diagnosis, many of them based on histopathological findings. Jellinger considers the following suggestions [188]:
1. National Institute of Aging (NIA) criteria assessing plaques (including diffuse plaques as neuritic-NPs) in neocortex and hippocampus per unit, corrected for age.
2. Criteria based in the semi-quantitative assessment of plaques and NFTs in neocortex and hippocampus.
3. CERAD criteria using the semi-quantitative NPs count adjusted for age, to which the clinical history of dementia is added to establish the probability of AD.
4. Topographic categorization of neuritic (neurofibrillary) changes, with 6 stages: entorhinal (1 and 2), hippocampal (3 and 4) and neocortical (5 and 6).
5. Quantitative criteria of the University of Washington, a modification of the NIA consensus criteria 1985.
6. The guides of NIA and Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association (NIA-RI) make a postmortem diagnosis of AD by combining the CERAD and Braak criteria. This leads us to establish a low probability (CERAD = 0-A, Braak = 1-2), medium probability (CERAD B, Braak 3-4) or high probability (CERAD C, Braak 5-6) that dementia is caused by AD.
7. VD/VCI cluster all the cases where cognitive compromise can be attributed to cerebrovascular disease (CVD) and it is greater than that expected for normal aging. VD would be a type located at the end of VCI, where cognitive compromise is severe enough to interfere with social and occupational activities.
VCI is a “continuum” emerging from the initial appearance of vascular risk factors, which generate CVD and then cerebrovascular damage, which according to its location causes different types of cognitive compromise. CVD includes arteriolosclerosis, atherosclerosis, amyloid angiopathy and CADASIL, while cerebrovascular damage relates to a brain lesion caused by CVD, which will depend on the size of the involved artery, i.e., lesion in large arteries and lesions in small arteries. In the first case, it can cause total occlusion and we will have an infarct, with a deficient clinical picture, generally motor, that along the months may evolve into cognitive impairment, which is known as post-VCD dementia (or post-stroke dementia). In the case of cerebrovascular damage caused by lesions of small arteries, we have two probable settings, which finish in the classic lacunar state syndromes and Binswanger syndrome, depending on complete or incomplete artery occlusion. Thus, a pathological examination is important for, first, confirming or detecting cerebral vascular damage, especially for lesions that cannot be detected by neuroimaging techniques (small infarcts, selective neuronal loss, microinfarcts); second, confirming or identifying the type of underlying CVD (arteriolosclerosis, amyloid angiopathy, etc.); and third, to assess the presence or matching extension of an AD-type pathology. Jellinger proposes a combination of autopsy-proven AD and multiple lacunar infarcts or lesions causing cerebrovascular damage in cortex, basal ganglia, thalamus, hippocampus and white matter, with approximately 30-50 mL of infarcted cerebral volume.
Due to the difficulty to make available technological devices (MRI with more Tesla units, Pittsburgh compound B, etc.) and a histopathology in most of the cases, an operating definition for mixed dementia is recommended to use it in a stepped way as follows:
1. Carrier of cardiovascular and cerebrovascular risk factors, either known or not by the patient at the time of initial evaluation.
2. Defined diagnosis of dementia, without considering the neuropsychological phenotype.
3. MR neuroimaging with 1.5 Tesla or greater demonstrating vascular lesions other than:
• Lobar infarct or hemorrhage
• Strategic lesions (e.g.: thalamic)
• Extensive vascular lesions (Fazekas scale 3)
• Age, which has a greater importance in the pathophysiology of both diseases. The detection at early stages, at a younger age, has a greater relevance as after the age of 85 most of the cases are defined and advanced, and even the cases of degenerative dementia will already have a not negligible “lesion burden”. On the other hand, hereditary forms of Alzheimer’s disease have an early onset. For example, the mean age at onset is 46.8 years (range: 36- 42 years) with an average duration of up to 8 years with a fatal outcome in patients with dominant E280A mutation of the presenilin-1 gene present in Antioquia (Colombia). These cases are characterized by the presence of minimal cerebrovascular lesions, in contrast to forms of late-onset AD [205].
Strategies to Prevent Vascular Cognitive Impairment
There are several therapeutic interventions that can prevent, delay the onset or slow down the progression of vascular cognitive impairment, which are described in Table 4 [206-208]:
| Interventions |
Evidence quality (GRADE System) |
Strategy recommendations |
| Lifestyle factors |
| Smoking discontinuation |
B1 |
Reasonable |
| Moderate alcohol intake |
B1 |
It can be reasonable |
| Weight control |
B1 |
It can be reasonable |
| Physical activity |
A1 |
Advisable |
| Mediterranean diet |
B1 |
Advisable |
| Physiological factors |
| Treatment of hypertension |
A1 |
Advisable |
| Treatment of hyperglycemia |
B1 |
It can be reasonable |
| Treatment of hypercholesterolemia |
B1 |
It can be reasonable |
| Other interventions |
| Vitamin supplements |
A2 |
Not demonstrated |
| Cognitive stimulation |
B1 |
Advisable |
| Multimodal intervention |
B1 |
Advisable |
Table 4: Strategies for therapeutic intervention in vascular cognitive impairment.
The main impact on the modification of natural history of cognitive alterations related to cerebrovascular disorders is achieved when therapeutic interventions are applied in the early stages of cognitive impairment [209], but especially when they are conducted before VCI develops. These measures not only help to prevent VCI but also have favorable results in neurodegenerative disorders such as Alzheimer’s disease.
Health care efforts should be focused on primary preventive measures, including the modification of known vascular risk factors [206,207,210,211]. The direct impact on the improvement of cognitive functions through the appropriate control of smoking, hypertension [212,213] and the Mediterranean diet [60-63] is especially relevant.
Different studies and systematic reviews have demonstrated that cognitive training is an effective way to objectively improve the cognitive function. A Cochrane review in 2012 [214] points out that cognitive stimulation programs demonstrated benefits in the treatment of patients with mild MCI and moderate dementia. A relation between mental activity developed early, in the middle and late life, and a significant reduction of dementia has been observed in the advanced age (OR: 0.54). A multivariate approach to reducing risk for cognitive decline was tested in the FINGER study (Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability) [215]. Authors found overall that cognitive performance improved significantly with a multicomponent lifestyle intervention involving physical activity, nutritional guidance, cognitive training, and social activities, as well as management of vascular risk factors.
Recommendations
In the absence of a disease-modifying treatment or cure for CVI, reducing the risk of developing vascular dementia takes on added importance. There is sufficiently strong evidence to conclude that a healthy diet, management of modifiable vascular risk factors (diabetes, obesity, smoking, and hypertension) and lifelong learning and cognitive training may reduce the risk of cognitive decline and may reduce the risk of dementia.
Pharmacological Treatment
The main randomized controlled trials in patients with VCI treated with pharmacologic interventions show some benefit for improving cognition. However, none have received regulatory approval particularly because the lower effect sizes compared to AD. Considered the previous aspects, the main drugs are briefly reviewed [216].
Donepezil
It is a specific and reversible acetylcholinesterase (AChE) inhibitor that increases acetylcholine (ACh) levels. Several studies using donepezil for vascular dementia have been published. Two randomized, double blind, controlled studies in 1219 patients with possible or probable vascular cognitive impairment should be mentioned; in them, 5 or 10 mg daily of the drug were administered for 6 months, compared to placebo. Significant benefits were found in cognition, global function and functionality in activities of daily life in patients receiving donepezil, thereby inferring its effectiveness [217,218].
Recommendation Grade: A1
Rivastigmine
This pseudo-irreversible inhibitor of acetylcholinesterase and butyrylcholinesterase, facilitates cholinergic neurotransmission by slowing degradation of acetylcholine released by functionally intact cholinergic neurons.
A double-blind, randomized clinical trial assessed the effectiveness of rivastigmine treatment (3-12 mg/day) compared to placebo in patients with vascular dementia for 6 months. The results did not provide consistent efficacy [219]. The other trial compared rivastigmine (9 mg/day) with placebo in patients with mild cognitive impairment for 6 months of treatment, providing some benefit in the executive functioning [220].
Recommendation Grade: B2
Galantamine
It is a competitive, reversible acetylcholinesterase inhibitor. It shows allosteric modulation of the nicotinic receptor (nAChR), and bonds to nAChR in a place other than acetylcholine (ACh). When it merges simultaneously with ACh at their respective binding sites, the presynaptic nicotinic receptor becomes more sensitive to ACh and amplifies its response.
Galantamine has been assessed in two clinical trials crucial to know the benefits of this drug. A double-blind, randomized clinical trial used galantamine (24 mg/day) in 592 patients with “pure” vascular dementia as “mixed” vascular dementia for 6 months of treatment, showing a benefit over placebo in cognition, global assessment, activities of daily life and behavioral symptoms [221]. The other trial in 788 patients diagnosed with “pure” vascular dementia assessed the effectiveness of galantamine treatment with 24 mg/day, compared to placebo for 26 weeks. The results showed a benefit in cognition and executive functions with galantamine [222,223].
Recommendation Grade: B1
Memantine
It is a non-competitive, voltage-dependent, antagonist with low/ moderate affinity for NMDA (N-methyl-D-aspartate) receptors; it blocks the excitotoxic effects of pathologically increased glutamate tonic levels that may cause neuronal dysfunction.
Two clinical trials compared memantine (20 mg/day) to placebo in mild to moderate vascular dementia for 28 weeks. Data analysis showed an improvement in cognitive functions, but not in the global clinical impression nor in the activities of daily life, in those treated with memantine [224,225].
Recommendation Grade: B2
Nimodipine
It binds to L-type calcium receptors and reduces the number of open channels transporting calcium ions through cell membrane. It has a vasodilating action on arterioles and can modulate other calcium dependent processes such as the release of acetylcholine. The binding sites are in neurons and cerebrovascular cells, and their action can influence both neural conduction and brain blood flow.
A Cochrane 2002 review analyzed three studies using nimodipine (90 mg/day) between 12 and 24 weeks in patients with vascular dementia, and a benefit was found in some cognition and global assessment scales [226] A multicenter, double-blind, randomized study comparing nimodipine (90 mg/day) to placebo for the treatment of subcortical vascular dementia for 52 weeks was published, without finding a benefit in the cognition scales [227].
Recommendation Grade: B2
Citicoline
It stimulates the biosynthesis of structural phospholipids in the neuronal membrane. It inhibits the activation of certain phospholipases (A, A2, C and D) and preserves the neuronal energy reserve, inhibiting apoptosis and stimulating the synthesis of acetylcholine. It improves the brain function by interacting with several neurotransmitters and receptors, as well as due to its vasoactive and antibinding properties.
A systematic Cochrane 2005 review included 14 clinical trials; the effects of citicoline on the treatment of cognitive, emotional and behavioral deficits associated with chronic cerebral disorders in older people were analyzed. There were difficulties due to heterogeneity of the articles, but even with these technical difficulties there was evidence of certain benefit for memory, behavior and global clinical impression in the medium and short term, especially in patients with cognitive deficits associated with cerebrovascular disorders [228]. Subsequently, a randomized, open-label study comparing citicoline to placebo for 12 months was published to assess the effectiveness of citicoline in preventing post-stroke cognitive impairment in patients who had a first ischemic stroke event; benefits in executive function and guidance were found [229,230]. Simultaneously, the IDEALE study was published, comparing oral citicoline (1000 mg/day) to placebo for mild VCI for 9 months. The results obtained showed a slight but non-significant difference in the geriatric depression scale score, and also that MMSE score remained unaltered over the time [17,231]. A two-year treatment and follow-up study with citicoline published in 2016 having as objective to measure the quality of life and cognitive status in patients who had suffered a first stroke demonstrated that citicoline (1 g/day) is associated with an improved quality of life (EuroQol-5D) in both elderly and young patients, and significantly improved the cognitive status (p = 0.005) after two years of treatment, following a first stroke. In addition, only the citicoline treated group (vs. standard measures) sustained cognitive improvement after the first year of treatment [232].
Recommendation Grade: B2
EGB 761 (Ginkgo biloba).
Ginkgo biloba has antioxidant, vasoactive and neurotransmittermodulating properties.
In the Cochrane 2002 review, it was concluded that the benefit of Ginkgo biloba (unspecified dose) for people with dementia or cognitive impairment is inconsistent and unreliable. Subsequently, a multicenter, double-blind, randomized study was conducted to demonstrate the effectiveness of Ginkgo biloba (240 mg/day) compared to placebo in 410 patients with mildmoderate dementia (vascular dementia or Alzheimer’s disease) for 24 weeks. The results showed a benefit of using Ginkgo biloba compared to placebo in cognition, psychopathology, functional measures and quality of life for patients and caragivers [233].
Recommendation Grade: B2
Cerebrolysin
It has neurotrophic mechanisms such as antiapoptopic activity, modulation of anti-inflammatory response, reduction of free radicals, modulation of CDK5 and GSK3β activity, neuroplasticity and neurogenesis.
In a randomized, double-blind, prospective clinical trial conducted with cerebrolysin in 242 patients over 50 years of age diagnosed with mild-moderate vascular dementia, 20 mL intravenous cerebrolysin were administered once daily for 24 weeks, comparing to placebo. The cerebrolysin treatment showed that cognitive function, overall function, daily life activities and executive functions improved significantly [234,235]. Subsequently, a Cochrane 2013 review was published and analyzed 6 studies using 10-30 mL intravenous cerebrolysin daily with a variable duration (age range: 60-71 years) in patients diagnosed with mild-moderate vascular dementia, and a benefit in some scales of cognition and global assessment was found [236].
Recommendation Grade: C2
20887
References
- GBD 2015 Mortality and Causes of Death Collaborators (2016) Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study of 2015. Lancet 388: 1459-1554.
- Prince M, Wimo A, Guerchet M, Ali G, Wu Y, et al. (2015) Informe Mundial sobre el Alzheimer 2015. Las consecuencias de la demencia análisis de prevalencia, incidencia, coste y tendencias. Londres: Alzheimer’s Disease International.
- De Carvalho JJ, Alves MB, Viana GA, Machado CB, Dos Santos BF, et al. (2011) Stroke epidemiology, patterns of management, and outcomes in Fortaleza, Brazil: a hospital-based multicenter prospective study. Stroke 42: 3341-3346.
- Porcello Marrone LC, Diogo LP, De Oliveira FM, Trentin S, Scalco RS, et al. (2013) Risk factors among stroke subtypes in Brazil. J Stroke Cerebrovasc Dis 22: 32-35.
- Saposnik G, Del Brutto OH (2003) Stroke in South America: A systematic review of incidence, prevalence, and stroke subtypes. Stroke 34: 2103-2107.
- Marquez-Romero JM, Arauz A, Gongora-Rivera F, Barinagarrementeria F, Cantu C (2015) The burden of stroke in Mexico. Int J Stroke 10: 251-252.
- Bahit MC, Coppola ML, Riccio PM, Cipriano LE, Roth GA, et al. (2016) First-ever stroke and transient ischemic attack incidence and 30-day case-fatality rates in a population-based study in Argentina. Stroke 47: 1640-1642.
- Lavados PM, Sacks C, Prina L, Escobar A, Tossi C, et al. (2005) Incidence, 30-day case-fatality rate, and prognosis of stroke in Iquique, Chile: a 2-year community-based prospective study (PISCIS project). Lancet. 365: 2206-2215.
- Cabral NL, Cougo-Pinto PT, Magalhaes PS, Longo AL, Moro CH, et al. (2016) Trends of stroke incidence from 1995 to 2013 in Joinville, Brazil. Neuroepidemiology 46: 273-281.
- Cantu-Brito C, Majersik JJ, Sanchez BN, Ruano A, Quiñones G, et al. (2010) Hospitalized stroke surveillance in the community of Durango, Mexico: the brain attack surveillance in Durango study. Stroke 41: 878-884.
- Cantu-Brito C, Majersik JJ, Sanchez BN, Ruano A, Becerra-Mendoza D, et al. (2011) Door-to-door capture of incident and prevalent stroke cases in Durango, Mexico: the brain attack surveillance in Durango study. Stroke 42: 601-606.
- Melcon CM, Melcon MO (2006) Prevalence of stroke in an Argentine community. Neuroepidemiology 27: 81-88.
- Thrift AG, Cadilhac DA, Thayabaranathan T, Howard G, Howard VJ, et al. (2014) Global stroke statistics. Int J Stroke 9: 6-18.
- Nitrini R, Bottino CM, Albala C, Custodio Capuñay NS (2009) Prevalence of dementia in Latin America: a collaborative study of population-based cohorts. Inter Psychogeriatr 21: 622–630.
- Rizzi L, Rosset I, Roriz CM (2014) Global epidemiology of dementia: Alzheimer's and vascular types. Biomed Res Int 2014: 908915.
- Arauz A, Rodríguez AY, Sosa AL, Chávez M, Paz F, et al. (2014) Vascular cognitive disorders and depression after first-ever stroke: the Fogarty-Mexico Stroke Cohort. Cerebrovasc Dis 38: 284-289.
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. (2005) Global prevalence of dementia: A Delphi consensus study. Lancet 3667: 2112-2117.
- Alonso CP, Oxman AD, Moberg J, Brignardello PR, Akl EA, et al. (2016) GRADE Evidence to Decision (EtD) frameworks: A systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines BMJ. 353: i2089.
- Reisberg B, Ferris SH, Kluger A, Franssen E, Wegiel J, et al. (2008) Mild cognitive impairment (MCI): A historical perspective. Int Psychogeriatr 20: 18-31.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, et al. (1999) Mild cognitive impairment: Clinical characterization and outcomes. Arch Neurol 56: 303-308.
- Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE, et al. (2009) Mild cognitive impairment: Ten years later. Arch Neurol 66: 1447-1455.
- Sachdev P, Kalaria R, O’Brien J, Skoog I, Alladi S, et al. (2014) Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis Assoc Disord 28: 206–218.
- American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders (5th edn) (DSM-5). American Psychiatric Publishing, Washington, DC, USA.
- Román GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, et al. (1993) AIREN International Workshop. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43: 250-260.
- Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, et al. (1992) Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer's Disease Diagnostic and Treatment Centers. Neurology 42: 473-80.
- Roman GC (1987) Senile dementia of the Binswangeer type: A vascular from of dementia in the elderly. JAMA 258: 1782–1788.
- Carrera E, Bogousslavsky J (2006) The thalamus and behavior: Effects of anatomically distinct strokes. Neurology 66: 1817-1823.
- Zekry D, Duyckaerts C, Belmin J, Geoffre C, Herrmann F, et al. (2003) The vascular lesions in vascular and mixed dementia: the weight of functional neuroanatomy. Neurobiol Aging 24: 213-219.
- Del Ser T, Bermejo F, Portera A, Arredondo JM, Bouras C, et al. (1990) Vascular dementia: A clinicopathological study. J Neurol Sci 96: 1-17.
- Tomlinson BE, Blessed G, Roth M (1970) Observations on the brains of demented old people. J Neurol Sci 11: 205-242.
- Brun A (1994) Pathology and pathophysiology of cerebrovascular dementia: Pure subgroups of obstructive and hypoperfusive etiology. Dementia 5: 145-147.
- Erkinjuntti T, Haltia M, Palo J, Sulkava R, Paetau A (1988) Accuracy of the clinical diagnosis of vascular dementia: a prospective clinical and post-mortem neuropathological study. J Neurol Neurosurg Psychiatry 51: 1037-1044.
- Skrobot OA, O'Brien J, Black S, Chen C, DeCarli C, et al. (2017) The Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement 13: 624-633.
- Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, et al. (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226: 75-80.
- Lanna ME, Alves CE, Sudo FK, Alves G, Valente L, et al. (2012) Cognitive disconnective syndrome by single strategic strokes in vascular dementia. J Neurol Sci 322: 176-183.
- Catani M, Mesulam M (2008) The arcuate fasciculus and the disconnection theme in language and aphasia: history and current state. Cortex 44: 953-961.
- Jellinger KA (2013) Pathology and pathogenesis of vascular cognitive impairment – A critical update. Front Aging Neurosci 5: 17.
- Van der Flier WM, Cordonnier C (2012) Microbleeds in vascular dementia: Clinical aspects. Exp Gerontol 47: 853-857.
- Rincon F, Wrigth CB (2013) Vascular cognitive impairment. Current Opinion in Neurology 26: 29-36.
- Chui HC (2007) Subcortical ischemic vascular dementia. Neurol Clin 25: 717-740.
- Leyds D, Hénon H, Mackowiak-Cordoliania MA, Pasquier F (2005) Poststroke dementia. Lancet Neurol 4: 752-759.
- Kim KW, Youn JC, Han MK, Paik NJ, Lee TJ, et al. (2008) Lack of association between apolipoprotein E polymorphism and vascular dementia in Koreans. J Geriatr Psychiatry Neurol 21: 12-17.
- Kuller LH, Lopez OL, Jagust WJ, Becker JT, DeKosky ST, et al. (2005) Determinants of vascular dementia in the cardiovascular health cognition study. Neurology 64: 1548-1552.
- Chabriat H, Bousser MG (2003) CADASIL: Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Adv Neurol 92: 147-150.
- Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M (2004) Long term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain 127: 2533- 2539.
- Cherubini A, Martin A, Andres LC, Di Iorio A, Lamponi M, et al. (2005) Vitamin E levels, cognitive impairment and dementia in older persons: the InCHIANTI study. Neurobiol Aging 26: 987-994.
- Jama JW, Launer LJ, Witteman JC, Den Breeijen JH, Breteler MM, et al. (1996) Dietary antioxidants and cognitive function in a population-based sample of older persons: the Rotterdam study. Am J Epidemiol 144: 275–280.
- Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS (2006) Associations of vegetable and fruit consumption with age-related cognitive change. Neurology 67: 1370–1376.
- Kang JH, Grodstein F (2008) Plasma carotenoids and tocopherols and cognitive function: a prospective study. Neurobiol Aging 29: 1394–1403.
- Kang JH, Cook NR, Manson JE, Buring JE, Albert CM, et al. (2009) Vitamin E, vitamin C, beta carotene, and cognitive function among women with or at risk of cardiovascular disease: the women's anti-oxidant and cardiovascular study. Circulation 119: 2772–2780.
- Heart Protection Study Collaborative Group (2002) MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 360: 23-33.
- Bourre JM (2006) Effects of nutrients (in food) on the structure and function of the nervous system: An update on dietary requirements for brain, part 2: Macronutrients. J Nutr Health Aging 10: 386-399.
- Chew EY, Clemons TE, Agrón E, Launer LJ, Grodstein F, et al. (2015) Effect of omega-3 fatty acids, lutein/zeaxanthin, or other nutrient supplementation on cognitive function. The AREDS2 randominze clinical trial. JAMA 314: 791-801.
- Kalmijn S, Feskens EJ, Launer LJ, Kromhout D (1997) Polyunsaturated fatty acids, antioxidants, and cognitive function in very old men. Am J Epidemiol 145: 33-41.
- Heude B, Ducimetière P, Berr C (2003) Cognitive decline and fatty acid composition of erythrocyte membranes: the EVA Study. Am J Clin Nutr 77: 803-808.
- Beydoun MA, Kaufman JS, Satia JA, Rosamond W, Folsom AR (2007) Plasma n-3 fatty acids and the risk of cognitive decline in older adults: the Atherosclerosis Risk in Communities Study. Am J Clin Nutr 85: 1103-1111.
- Kalmijn S, van Boxtel MP, Ocke M, Verschuren WM, Kromhout D, et al. (2004) Dietary intake of fatty acids and fish in relation to cognitive performance at middle age. Neurology 62: 275-280.
- Van de Rest O, Spiro A, Krall-Kaye E, Geleijnse JM, De Groot LC, et al. (2009) Intakes of (n-3) fatty acids and fatty fish are not associated with cognitive performance and 6-year cognitive change in men participating in the Veterans Affairs Normative Aging Study. J Nutr 139: 2329-2336.
- Scarmeas N, Stern Y, Mayeux R, Manly JJ, Schupf N, et al. (2009) Mediterranean diet and mild cognitive impairment. Arch Neurol 66: 216-225.
- Féart C, Samieri C, Rondeau V, Amieva H, Portet F, et al. (2009) Adherence to a Mediterranean diet, cognitive decline and risk of dementia. JAMA. 302: 638-648.
- Tangney CC, Kwasny MJ, Li H, Wilson RS, Evans DA, et al. (2011) Adherence to a Mediterranean diet and age-related cognitive decline a randomized clinical trial. Am J Clinn Nutr 93: 601-607.
- Valls-Pedret C, Sala-Vila A, Serra-Mir M, Corella D, De la Torre R, et al. (2015) Mediterranean diet and age-related cognitive decline: a randomized clinical trial. JAMA Intern Med 175: 1094-1103.
- Gustafson DR, Karlsson C, Skoog I, Rosengren L, Lissner L, et al. (2007) Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J Intern Med 262: 643-650.
- Beydoun MA, Beydoun HA, Wang Y (2008) Obesity and central obesity as risk factors for incident dementia and its subtypes: a systematic review and meta-analysis. Obes Rev 9: 204-218.
- Fitzpatrick AL, Kuller LH, Lopez OL, Diehr P, O'Meara ES, et al. (2009) Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch Neurol 66: 336-342.
- Stewart R, Masaki K, Xue QL, Peila R, Petrovitch H, et al. (2005) A 32-year prospective study of change in body weight and incident dementia: The Honolulu-Asia Aging Study. Arch Neurol 62: 55-60.
- Wolf PA, Beiser A, Elias MF, Au R, Vasan RS, et al. (2007) Relation of obesity to cognitive function: importance of central obesity and synergistic influence of concomitant hypertension: The Framingham Heart Study. Curr Alzheimer Res 4: 111–116.
- Román GC (2011) Pathogenesis of cerebral small-vessel disease in obstructive sleep apnea. In: Culebras A, (ed) Sleep, Stroke, and Cardiovascular Disease, Chapter 8. Cambridge University Press, New York, USA. pp: 97-113.
- Sink KM, Espeland MA, Castro CM, Church T, Cohen R, et al. (2015) LIFE Study Investigators. Effect of a 24-month physical activity intervention vs healt education on cognitive outcomes in sedentary older adults. The LIFE Randomized Trial. JAMA 34: 781-790.
- Gill SD, Seltz DP (2015) Lifestyles and cognitive health. What older individuals can do to optimize cognitive outcomes. Editorial. JAMA 314: 774-775.
- Weuve J, Kang JH, Manson JE, Breteler MM, Ware JH, et al. (2004) Physical activity, including walking, and cognitive function in older women. JAMA 292: 1454–1461.
- Sturman MT, Morris MC, Mendes de Leon CF, Bienias JL, Wilson RS, et al. (2005) Physical activity, cognitive activity, and cognitive decline in a biracial community population. Arch Neurol 62: 1750-1754.
- Larson EB, Wang L, Bowen JD, McCormick WC, Teri L, et al. (2006) Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 144: 73-81.
- Ravaglia G, Forti P, Lucicesare A, Pisacane N, Rietti E, et al. (2008) Physical activity and dementia risk in the elderly: findings from a prospective Italian study. Neurology 70: 1786-1794.
- Angevaren M, Aufdemkampe G, Verhaar HJ, Aleman A, Vanhees L (2008) Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst Rev 16: CD005381.
- Soumaré A, Tavernier B, Alpérovitch A, Tzourio C, Elbaz A (2009) A cross-sectional and longitudinal study of the relationship between walking speed and cognitive function in community-dwelling elderly people. J Gerontol A Biol Sci Med Sci 64: 1058-1065.
- Yaffe K, Fiocco AJ, Lindquist K, Vittinghoff E, Simonsick EM, et al. (2009) Predictors of maintaining cognitive function in older adults: the Health ABC study. Neurology 72: 2029-2035.
- Verghese J, Cuiling W, Katz MJ, Sanders A, Lipton RB (2009) Leisure activities and risk of vascular cognitive impairment in older adults. J Geriatr Psychiatry Neurol 22: 110-118.
- Liu AT, Eng JJ, Boyd LA, Jacova C, Davis JC, et al. (2010) Promotion of the mind through exercise (PROMoTE): a proof-of-concept randomized controlled trial of aerobic exercise training in older adults with vascular cognitive impairment. BMC Neurol 10: 14.
- Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, et al. (2010) Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Arch Neurol 67: 71–79.
- Sofi F, Valecchi D, Bacci D, Abbate R, Gensini GF, et al. (2011) Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. J Intern Med 269: 107-117.
- Kalmijn S, Van Boxtel MP, Verschuren MW, Jolles J, Launer LJ (2002) Cigarette smoking and alcohol consumption in relation to cognitive performance in middle age. Am J Epidemiol 156: 936-944.
- Anstey KJ, Von Sanden C, Salim A, O'Kearney R (2007) Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. Am J Epidemiol 166: 367-378.
- Sabia S, Marmot M, Dufouil C, Singh MA (2008) Smoking history and cognitive function in middle age from the Whitehall II study. Arch Intern Med 168: 1165-1173.
- Poorthuis RB, Goriounova NA, Couey JJ, Mansvelder HD (2009) Nicotinic actions on neuronal networks for cognition: general principles and long-term consequences. Biochem Pharmacol 78: 668-676.
- Sabia S, Elbaz A, Dugravot A, Head J, Shipley M, et al. (2012) Impact of smoking on cognitive decline in early old age: the Whitehall II cohort study. Arch Gen Psychiatry 69: 627-635.
- North TL, Palmer TM, Lewis SJ, Cooper R, Power C, et al. (2015) Effect of smoking on physical and cognitive capability in later life: a multicohort study using observational and genetic approaches. BMJ Open 5: e008393.
- Elias PK, Elias MF, D'Agostino RB, Silbershatz H, Wolf PA (1999) Alcohol consumption and cognitive performance in the Framingham Heart Study. Am J Epidemiol 150: 580-589.
- Ganguli M, Vander Bilt J, Saxton JA, Shen C, Dodge HH (2005) Alcohol consumption and cognitive function in late life: a longitudinal community study. Neurology 65: 1210–1217.
- Stampfer MJ, Kang JH, Chen J, Cherry R, Grodstein F (2005) Effects of moderate alcohol consumption on cognitive function in women. N Engl J Med 352: 245-253.
- Stott DJ, Falconer A, Kerr GD, Murray HM, Trompet S, et al. (2008) Does low to moderate alcohol intake protect against cognitive decline in older people? J Am Geriatr Soc 56: 2217-2224.
- Kim JW, Lee DY, Lee BCH, Jung MH, Kim H, et al. (2012) Alcohol and cognition in the elderly: A review. Psychiatry Investig 9: 8-16.
- Bernardin F, Maheut-Bosser A, Paille F (2014) Cognitive impairments in alcohol-dependent subjects. Front Psychiatry 5: 78.
- Sameer J, Marshall EJ, Smith ID (2014) Alcohol and cognitive impairment. BJ Psych Advances 20: 304-313.
- Yaffe K, Blackwell T, Gore R, Sands L, Reus V, et al. (1999) Depressive symptoms and cognitive decline in nondemented elderly women: a prospective study. Arch Gen Psychiatry 56: 425-430.
- Ganguli M, Du Y, Dodge HH, Ratcliff GG, Chang CC (2006) Depressive symptoms and cognitive decline in late life: a prospective epidemiological study. Arch Gen Psychiatry 63: 153-160.
- Hammar A, Ardal G (2009) Cognitive functioning in major depression - A summary. Frontiers in Human Neuoscience 3: 26.
- Godin O, Dufouil C, Ritchie K, Dartigues JF, Tzourio C, et al. (2007) Depressive symptoms, major depressive episode and cognition in the elderly: the three-city study. Neuroepidemiology 28: 101-108.
- Papazacharias A, Nardini M (2012) The relationship between depression and cognitive deficits. Psychiatria Danubina 24: S179-S182.
- Barnes DE, Alexopoulos GS, Lopez OL, Williamson JD, Yaffe K (2006) Depressive symptoms, vascular disease, and mild cognitive impairment: findings from the Cardiovascular Health Study. Arch Gen Psychiatry 63: 273-279.
- Cankurtaran M, Yavuz BB, Cankurtaran ES, Halil M, Ulger Z, et al. (2008) Risk factors and type of dementia: Vascular or Alzheimer?. Arch Gerontol Geriatr 47: 25-34.
- Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, et al. (2004) Inflammatory proteins in plasma and the risk of dementia: the Rotterdam study. Arch Neurol 61: 668-672.
- Schmidt R, Schmidt H, Curb JD, Masaki K, White LR, et al. (2002) Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann Neurol 52: 168– 174.
- Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, et al. (2007) Blood inflammatory markers and risk of dementia: the Conselice Study of Brain Aging. Neurobiol Aging 28: 1810-1820.
- Seliger SL, Siscovick DS, Stehman-Breen CO, Gillen DL, Fitzpatrick A, et al. (2004) Moderate renal impairment and risk of dementia among older adults: the Cardiovascular Health Cognition Study. J Am Soc Nephrol 15: 1904-1911.
- Etgen T, Sander D, Chonchol M, Briesenick C, Poppert H, et al. (2009) Chronic kidney disease is associated with incident cognitive impairment in the elderly: the INVADE study. Nephrol Dial Transplant 24: 3144-3150
- Kurella M, Yaffe K, Shlipak MG, Wenger NK, Chertow GM (2005) Chronic kidney disease and cognitive impairment in menopausal women. Am J Kidney Dis 45: 66–76.
- Elias MF, Elias PK, Seliger SL, Narsipur SS, Dore GA, et al. (2009) Chronic kidney disease, creatinine and cognitive functioning. Nephrol Dial Transplant 24: 2446–2452.
- Int Veld BA, Ruitenberg A, Hofman A, Stricker BH, Breteler MM (2001) Antihypertensive drugs and incidence of dementia: The Rotterdam Study. Neurobiol Aging 22: 407-412.
- Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, et al. (2000) Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol Aging 21: 49-55.
- Skoog I, Nilsson L, Persson G, Palmertz B, Andreasson LA, et al. (1996) 15-year longitudinal study of blood pressure and dementia. Lancet 347: 1141-1145.
- Cheng G, Huang C, Deng H, Wang H (2012) Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern Med J 42: 484–491.
- McCrimmon RJ, Ryan CM, Frier BM (2012) Diabetes and cognitive dysfunction. Lancet 379: 2291-2299.
- Saedi E, Gheini MR, Faiz F, Arami MA (2016) Diabetes mellitus and cognitive impairments. World J Diabetes 7: 412-422.
- Anstey KJ, Lipnicki DM, Low LF (2008) Cholesterol as a risk factor for dementia and cognitive decline: A systematic review of prospective studies with meta-analysis. Am J Geriatr Psychiatry 16: 343-354.
- Richardson K, Schoen M, French B, Umscheid C, Mitchell M, et al. (2013) Statins and cognitive function: A systematic review. Ann Intern Med 159: 688–697.
- Feinkohl I, Keller M, Robertson CM, Morling JR, Williamson RM, et al. (2013) Clinical and subclinical macrovascular disease as predictors of cognitive decline in older patients with type 2 diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes Care 36: 2779-2786.
- Hajjar I, Goldstein FC, Martin GS, Quyyumi AA (2016) Roles of arterial stiffness and blood pressure in hypertension-associated cognitive decline in healthy adults. Hypertension 67: 171-175.
- Rosano C, Naydeck B, Kuller LH, Longstreth WT Jr, Newman AB (2005) Coronary artery calcium: associations with brain magnetic resonance imaging abnormalities and cognitive status. J Am Geriatr Soc 53: 609-615.
- Vidal JS, Sigurdsson S, Jonsdottir MK, Eiriksdottir G, Thorgeirsson G, et al. (2010) Coronary artery calcium, brain function and structure: the AGES-Reykjavik Study. Stroke 41: 891–897.
- Laurin D, Masaki KH, White LR, Launer LJ (2007) Ankle-to-brachial index and dementia: the Honolulu-Asia Aging Study. Circulation 116: 2269-2274.
- Newman AB, Fitzpatrick AL, Lopez O, Jackson S, Lyketsos C, et al. (2005) Dementia and Alzheimer's disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J Am Geriatr Soc 53: 1101–1107.
- Devore EE, Grodstein F, Duffy JF, Stampfer MJ, Czeisler CA, et al. (2014) Sleep duration in midlife and later life in relation to cognition. J Am Geriatr Soc 62: 1073-1081.
- Chang WP, Liu ME, Chang WC, Yang AC, Ku YC, et al. (2013) Sleep apnea and the risk of dementia: A population-based 5-year follow-up study in Taiwan. PLoS One 8: e78655.
- Wald DS, Law M, Morris JK (2002) Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. Br Med J 325: 1202–1206.
- Kalmijn S, Launer LJ, Lindemans J, Bots ML, Hofman A, et al. (1999) Total homocysteine and cognitive decline in a community-based sample of elderly subjects. The Rotterdam Study. Am J Epidemiol 150: 283-289.
- Quadri P, Fragiacomo C, Pezzati R, Zanda E, Forloni G, et al. (2004) Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia. Am J Clin Nutr 80: 114–122.
- Garrett KD, Browndyke JN, Whelihan W, Paul RH, DiCarlo M, et al. (2004) The neuropsychological profile of vascular cognitive impairment–no dementia: Comparisons to patients at risk for cerebrovascular disease and vascular dementia. Arch Clin Neuropsychol 19: 745-757.
- Nyenhuis DL, Gorelick PB, Geenen EJ, Smith CA, Gencheva E, et al. (2004) The pattern of neuropsychological deficits in vascular cognitive impairment-no dementia (vascular CIND). Clin Neuropsychol 18: 41-49.
- Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, et al. (2006) National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37: 2220-2241.
- Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, et al. (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53: 695-699.
- Pendlebury ST, Mariz J, Bull L, Mehta Z, Rothwell PM (2012) MoCA, ACE-R, and MMSE versus the National Institute of Neurological Disorders and Stroke-Canadian Stroke Network Vascular Cognitive Impairment Harmonization Standards Neuropsychological Battery after TIA and stroke. Stroke 43: 464-469.
- Aguilar NS, Mimenza AA, Palacios GA, Samudio CA, Gutiérrez GL, et al. (2017) Validez y confiabilidad del MoCA (Montreal Cognitive Assessment) para el tamizaje del deterioro cognoscitivo en México. Revista Colombiana de Psiquiatria.
- Gómez F, Zunzunegui M, Lord C, Alvarado B, García A (2013) Applicability of the MoCA-S test in populations with little education in Colombia. Int J Geriatr Psychiatry 28: 813-820.
- Zhou Y, Ortiz F, Nuñez C, Elashoff D, Woo E, et al. (2015) Use of the MoCA in Detecting Early Alzheimer's Disease in a Spanish-Speaking Population with Varied Levels of Education. Dement Geriatr Cogn Dis Extra 5: 85-95.
- Carew TG, Lamar M, Cloud BS, Grossman M, Libon DJ (1997) Impairment in category fluency in ischemic vascular dementia. Neuropsychology 11: 400-412.
- Lamar M, Price CC, Davis KL, Kaplan E, Libon DJ (2002) Capacity to maintain mental set in dementia. Neuropsychologia 40: 435-445.
- Alexander MP, Stuss DT, Shallice T, Picton TW, Gillingham S (2005) Impaired concentration due to frontal lobe damage from two distinct lesion sites. Neurology 65: 572-579.
- Eslinger PJ, Grattan LM (1994) Altered serial position learning after frontal lobe lesion. Neuropsychologia 32: 729–739.
- Dubois B, Slachevsky A, Litvan I, Pillon B (2000) The FAB: A Frontal Assessment Battery at bedside. Neurology 55: 1621-1626.
- Shulman KI (2000) Clock-drawing: Is it the ideal cognitive screening test?. Int J Geriatr Psychiatry 15: 548-561.
- Eknoyan D, Hurley RA, Taber KH (2012) The clock drawing task: common errors and functional neuroanatomy. J Neuropsychiatry Clin Neurosci 24: 260-265.
- Osterrieth PA (1944) Le test de copie d’une figure complexe. Arch Psychologie 30: 206-356.
- Franzen M, Haut M, Rankin E, Keefover R (1995) Empirical comparison of alternative forms of the Boston naming test. Clin Neuropsychol 9: 225-229.
- Suades-González E, Jódar-Vicente M, Pérdrix-Solàs D (2009) Memory deficit in patients with subcortical vascular cognitive impairment versus Alzheimer-type dementia: The sensitivity of the 'word list' subtest on the Wechsler Memory Scale-III. Rev Neurol 49: 623-629.
- O’Sullivan M, Morris RG, Markus HS (2005) Brief cognitive assessment for patients with cerebral small vessel disease. J Neurol Neurosurg Psychiatry. 76: 1140-1145.
- Delis DC, Kramer JH, Kaplan E, Ober BA (2000) California Verbal Learning Test – (2nd edn), Adult version. Manual. Psychological Corporation, San Antonio, Texas, USA.
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, et al. (2013) Standards for Reporting Vascular changes on nEuroimaging (STRIVE v1). Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: 822-838.
- Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, et al. (2001) European Task Force on Age-Related White Matter Changes. A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke 32: 1318–1322.
- Brown WR, Moody DM, Thore CR, Challa VR, Anstrom JA (2007) Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J Neurol Sci 257: 62-66.
- Hilal S, Sikking E, Shaik MA, Chan QL, Van Veluw SJ, et al. (2016) Cortical cerebral microinfarcts on 3T MRI: A novel marker of cerebrovascular disease. Neurology 87: 1583-1590.
- Zipfel GJ, Han H, Ford AL, Lee JM (2009) Cerebral amyloid angiopathy: progressive disruption of the neurovascular unit. Stroke 40: S16-S19.
- Marcus C, Mena E, Subramaniam RM (2014) Brain PET in the diagnosis of Alzheimer's disease. Clin Nucl Med 39: e413-e422.
- Moorhouse P, Rockwood K (2008) Vascular cognitive impairment: Current concepts and clinical developments. Lancet Neurol 7: 246-255.
- Zetterberg H, Mattsson N, Blennow K (2010) Cerebrospinal fluid analysis should be considered in patients with cognitive problems. Int J Alzheimers Dis 2010: 163065.
- Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, et al. (2001) Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett 297: 187-190.
- Ost M, Nylén K, Csajbok L, Ohrfelt AO, Tullberg M, et al. (2006) Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67: 1600-1604.
- Samgård K, Zetterberg H, Blennow K, Hansson O, Minthon L, et al. (2010) Cerebrospinal fluid total tau as a marker of Alzheimer's disease intensity. Int J Geriatr Psychiatry 25: 403-410.
- Abdalla B, Bishara B, William N, Muhamad O, Mustafa Y (2015) Candidate Biomarkers and CSF Profiles for Alzheimer’s Disease and CADASIL. International Journal of Brain and Cognitive Sciences 4: 15-27.
- Petzold A, Keir G, Warren J, Fox N, Rossor MN (2007) A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegener Dis 4: 185-194.
- Abdulle S, Mellgren A, Brew BJ, Cinque P, Hagberg L, et al. (2007) CSF neurofilament protein (NFL) -a marker of active HIV-related neurodegeneration. J Neurol 254: 1026-1032.
- Gisslen M, Hagberg L, Brew BJ, Cinque P, Price RW, et al. (2007) Elevated cerebrospinal fluid neurofilament light protein concentrations predict the development of AIDS dementia complex. J Infect Dis 195: 1774-1778.
- Wallin A, Sjögren M (2001) Cerebrospinal fluid cytoskeleton proteins in patients with subcortical white-matter dementia. Mech Ageing Dev 122: 1937-1949.
- Tarkowski E, Tullberg M, Fredman P, Wikkelsö C (2003) Correlation between intrathecal sulfatide and TNF-alpha levels in patients with vascular dementia. Dement Geriatr Cogn Disord 15: 207-211.
- Bjerke M, Zetterberg H, Edman A, Blennow K, Wallin A, et al. (2011) Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in Vascular Dementia and Alzheimer's Disease. J Alzheimers Dis 27: 665-676.
- Román GC (2015) MTHFR Gene Mutations: A potential marker of late-onset Alzheimer's disease?. J Alzheimers Dis 47: 323-327.
- Kling MA, Trojanowski JQ, Wolk DA, Lee VM, Arnold SE (2013) Vascular disease and dementias: Paradigm shifts to drive research in new directions. Alzheimers Dement 9: 76-92.
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, et al. (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42: 2672-2713.
- Casserly I, Topol E (2004) Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet 363: 1139–1146.
- Toledo JB, Toledo E, Weiner MW, Jack CR Jr, Jagust W, et al. (2012) Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 8: 483-489.
- Polidori MC, Pientka L, Mecocci P (2012) A review of the major vascular risk factors related to Alzheimer’s disease. J Alzheimers Dis 32: 521-530.
- Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, et al. (2010) Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology 75: 1195-1202.
- Helzner EP, Luchsinger JA, Scarmeas N, Cosentino S, Brickman AM, et al. (2009) Contribution of vascular risk factors to the progression in Alzheimer disease. Arch Neurol 66: 343–348.
- Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, et al. (2006) Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol 5: 735-741.
- Mielke MM, Rosenberg PB, Tschanz J, Cook L, Corcoran C, et al. (2007) Vascular factors predict rate of progression in Alzheimer disease. Neurology 69: 1850-1858.
- Qiu C, Xu W, Winblad B, Fratiglioni L (2010) Vascular risk profiles for dementia and Alzheimer’s disease in very old people: a population-based longitudinal study. J Alzheimers Dis 20: 293-300.
- Moreno CC, Mimenza AA, Aguilar NS, Alvarado ÁP, Gutiérrez GL, et al. (2015) Factores asociados a demencia mixta en comparación con la demencia tipo Alzheimer en adultos mayores mexicanos. Psicogeriatría 5: 125-130.
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, et al. (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 136: 2697-2706.
- Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA (2011) Microinfarct pathology, dementia, and cognitive systems. Stroke 42: 722-727.
- Bennett DA, Wilson RS, Arvanitakis Z, Boyle PA, De Toledo-Morrell L, et al. (2013) Selected findings from the religious orders study and rush memory and aging project. J Alzheimers Dis 33: S397-S403.
- Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, et al. (2012) Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS One. 7: e41636.
- Ladecola C (2010) The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 120: 287-296.
- Riekse RG, Leverenz JB, McCormick W, Bowen JD, Teri L, et al. (2004) Effect of vascular lesions on cognition in Alzheimer’s disease: a community-based study. J Am Geriatr Soc 52: 1442–1448.
- Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28: 202-208.
- Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: 723-738.
- Du AT, Schuff N, Chao LL, Kornak J, Ezekiel F, et al. (2005) White matter lesions are associated with cortical atrophy more than entorhinal and hippocampal atrophy. Neurobiol Aging 26: 553-559.
- Jellinger KA, Attems J (2007) Neuropathological evaluation of mixed dementia. J Neurol Sci 257: 80-87.
- Schmidt R, Schmidt H, Haybaeck J, Loitfelder M, Weis S, et al. (2011) Heterogeneity in age-related white matter changes. Acta Neuropathol 122: 171-185.
- Artero S, Tiemeier H, Prins ND, Sabatier R, Breteler MM, et al. (2004) Neuroanatomical localisation and clinical correlates of white matter lesions in the elderly. J Neurol Neurosurg Psychiatry 75: 1304-1308.
- Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, et al. (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119: 579-589.
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, et al. (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41: 479-486.
- Kirsch W, McAuley G, Holshouser B, Petersen F, Ayaz M, et al. (2009) Serial susceptibility weighted MRI measures brain iron and microbleeds in dementia. J Alzheimers Dis 17: 599-609.
- Cordonnier C, Van der Flier WM, Sluimer JD, Leys D, Barkhof F, et al. (2006) Prevalence and severity of microbleeds in a memory clinic setting. Neurology 66: 1356-1360.
- Lee DY, Fletcher E, Martinez O, Ortega M, Zozulya N, et al. (2009) Regional pattern of White matter microstructural changes in normal aging, MCI, and AD. Neurology 73: 1722-1728.
- Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, et al. (1999) Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. Am J Neuroradiol 20: 637-642.
- Tanaka A, Ueno Y, Nakayama Y, Takano K, Takebayashi S (1999) Small chronic hemorrhages and ischemic lesions in association with spontaneous intracerebral hematomas. Stroke 30: 1637-1642.
- Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, et al. (2008) Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 63: 72-80.
- Zarow C, Sitzer TE, Chui HC (2008) Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep 8: 363-370.
- World Health Organization (1993) The ICD-10 classification of mental and behavioural disorders: Diagnostic criteria for research. En The ICD-10 classification of mental and behavioural disorders: Diagnostic criteria for research. World Health Organization.
- Hachinski VS, Iliff LD, Zilkha E, Du Boulay GH, McAllister VL, et al. (1975) Cerebral blood flow in dementia. Arch Neurol 32: 632-637.
- Loeb C, Gandolfo C (1983) Diagnostic Evaluation of degenerative and vascular dementia. Stroke 14: 399-401.
- Gold G, Bouras C, Canuto A, Bergallo MF, Herrmann FR, et al. (2002) Clinicopathological validation study of four sets of clinical criteria for vascular dementia. AM J Psychiatry 159: 82-87.
- Moroney Jt, Bagiella E, Desmond DW, Hachinski VC, Molsa PK, et al. (1997) Meta-analysis of the Hachinski ischemic score in pathologically verified dementias. Neurology 49: 1096-1105.
- Lopera F, Ardilla A, Martínez A, Madrigal L, Arango-Viana JC, et al. (1997) Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 277: 793-799.
- Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, et al. (2001) Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology 56: 1683-1689.
- Gorelick PB, Nyenhuis D (2013) Understanding and treating vascular cognitive impairment. Continuum (Minneap Minn) 19: 425-437.
- Román GC, Boller F (2014) Vascular factors in neurodegenerative diseases: A path towards treatment and prevention. Funct Neurol 29: 85-86.
- Román G, Pascual B (2012) Contribution of neuroimaging to the diagnosis of Alzheimer's disease and vascular dementia. Arch Med Res 43: 671-676.
- Cukierman-Yaffe T, Gerstein HC, Williamson JD, Lazar RM, Lovato L, et al. (2009) Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular risk factors: the action to control cardiovascular risk in diabetes-memory in adiabetes (ACCORD-MIND) trial. Diabetes Care 32: 221-226.
- Román GC, Nash DT, Fillit H (2012) Translating current knowledge into dementia prevention. Alzheimer Dis Assoc Disord 26: 295-299.
- Reitz C, Tang MX, Manly J, Mayeux R, Luchsinger JA (2007) Hypertension and the risk of mild cognitive impairment. Arch Neurol 64: 1734-1740.
- Birns J, Kalra L (2009) Cognitive function and hypertension. J Hum Hypertens 23: 86-96.
- Woods B, Aguirre E, Spector AE, Orrell M (2012) Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev 2: CD005562.
- Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, et al. (2015) A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385: 2255-2263.
- Smith EE, Cieslak A, Barber P, Chen J, Chen YW, et al. (2017) Therapeutic Strategies and Drug Development for Vascular Cognitive Impairment. J Am Heart Assoc 6: e005568.
- Román GC, Wilkinson DG, Doody RS, Black SE, Salloway SP, et al. (2005) Donepezil in vascular dementia: combined analysis of two large-scale clinical trials. Dement Geriatr Cogn Disord 20: 338-344.
- Román GC (2014) Vascular Cognitive Disorders. In, Gabbard’s Treatments of Psychiatric Disorders. DSM-5 Edition. pp: 977-985.
- Ballard C, Sauter M, Scheltens P, He Y, Barkhof F, et al. (2008) Efficacy, safety and tolerability of rivastigmine capsules in patients with probable vascular dementia: the VantagE study. Curr Med Res Opin 24: 2561-2574.
- Narasimhalu K, Effendy S, Sim CH, Lee JM, Chen I, et al. (2010) A randomized controlled trial of rivastigmine in patients with cognitive impairment no dementia because of cerebrovascular disease. Acta Neurol Scand 121: 217-224.
- Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, et al. (2002) Efficacy of galantamine in probable vascular dementia and Alzheimer's disease combined with cerebrovascular disease: A randomised trial. Lancet 359: 1283-1290.
- Auchus AP, Brashear HR, Salloway S, Korczyn AD, De Deyn PP, et al. (2007) GAL-INT-26 Study Group. Galantamine treatment of vascular dementia: A randomized trial. Neurology 69: 448-458.
- Román G, Pascual B (2012) Demencia Vascular y Deterioro Cognitivo de Origen Vascular. Revista Neuropsicología, Neuropsiquiatría y Neurociencias 12: 203-213.
- Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F (2002) Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke 33: 1834-1839.
- Wilcock G, Mobius HJ, Stoffler A, MMM 500 group (2002) A double-blind, placebo-controlled multicentre study of memantine in mild to moderate vascular dementia (MMM500). Int Clin Psychopharmacol 17: 297-305.
- López-Arrieta JM, Birks J (2002) Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev 3: CD000147.
- Pantoni L, Del Ser T, Soglian AG, Amigoni S, Spadari G, et al. (2005) Efficacy and safety of nimodipine in subcortical vascular dementia: a randomized placebo-controlled trial. Stroke 36: 619-624.
- Fioravanti M, Yanagi M (2005) Cytidinediphosphocholine (CDP-choline) for cognitive and behavioural disturbances associated with chronic cerebral disorders in the elderly. Cochrane Database Syst Rev 2: CD000269.
- Alvarez-Sabín J, Ortega G, Jacas C, Santamarina E, Maisterra O, et al. (2013) Long-term treatment with citicoline may improve poststroke vascular cognitive impairment. Cerebrovasc Dis 35: 146-154.
- Mimenza-Alvarado A, Aguilar-Navarro S, Ávila-Funes JA (2013) Guía Práctica de Demencias para diagnóstico y tratamiento. México: Corporativo Intermédica (Editorial Corinter) pp. 61-92.
- Cotroneo AM, Castagna A, Putignano S, Lacava R, Fantò F, et al. (2013) Effectiveness and safety of citicoline in mild vascular cognitive impairment: the IDEALE study. Clin Interv Aging 8: 131-137.
- Alvarez-Sabín J, Santamarina E, Maisterra O, Jacas C, Molina C, et al. (2016) Long-term treatment with citicoline prevents cognitive decline and predicts a better quality of life after a first ischemic stroke. J. et al. Int J Mol Sci 17: 390.
- Herrschaft H, Nacu A, Likhachev S, Sholomov I, Hoerr R, et al. (2012) Ginkgo biloba extract EGb 761® in dementia with neuropsychiatric features: A randomised, placebo-controlled trial to confirm the efficacy and safety of a daily dose of 240 mg. J Psychiatr Res 46: 716-723.
- Guekht AB, Moessler H, Novak PH, Gusev IE (2011) Cerebrolysin in vascular dementia: improvement of clinical outcome in a randomized, double-blind, placebo-controlled multicenter trial. J Stroke Cerebrovasc Dis 20: 310-318.
- Pearce LA, McClure LA, Anderson DC, Jacova C, Sharma M, et al. (2014) Effects of long-term blood pressure lowering and dual antiplatelet treatment on cognitive function in patients with recent lacunar stroke: A secondary analysis from the SPS3 randomised trial. Lancet Neurol 13: 1177-1185.
- Chen N, Yang M, Guo J, Zhou M, Zhu C, et al. (2013) Cerebrolysin for vascular dementia. Cochrane Database Syst Rev 1: CD008900.
- Flicker L, Evans G (2001) Piracetam for dementia or cognitive impairment. Cochrane Database Syst Rev 2: CD001011.
- Frampton M, Harvey RJ, Kirchner V (2003) Propentofylline for dementia. Cochrane Database Syst Rev 2: CD002853.

