Keywords
Felodipine; Solid dispersion; Microcapsules; HPMC; Gelatin
Introduction
Felodipine is a poorly water-soluble drug and when administered orally in the crystalline form has poor bioavalability [1]. For poorly soluble drugs, dissolution is the rate-limiting step for the gastrointestinal absorption from solid dosage forms. Increasing the water solubility can improve bioavailability of drug and this improved form of drug can also be used for preparation of controlled release dosage form.
Numerous attempts have been made to modify the dissolution characteristic of drug to attain more rapid and complete absorption [2- 4]. Solid dispersion is one of the techniques which are originally used to enhance the dissolution rate of poorly water-soluble drugs using inert hydrophilic carriers [5]. The enhancement in the dissolution rate is obtained by one or a combination of the following mechanism: eutectic formation, increased surface area of the drug due to precipitation in the carrier, formation of true solid solution, improved wettability due to close contact with a hydrophilic carrier, precipitation as a metastable crystalline form or a decrease in substance crystallinity. The type of solid dispersion formed depends on both the carrier-drug combination and the method of manufacture [6].
Hydrophilic polymers like polyethylene glycols (PEG) [7-9]. Polyvinyl pyrrolidone (PVP) [10,11] and hydroxyl propyl methyl cellulose (HPMC) [12] are among the popular carriers commonly used to prepare solid dispersions. Being freely soluble in water, these polymers are mainly used as excipients, to enhance the dissolution rate of drugs [13,14]. Pluronic F-68 is being used as a newer material for preparation of solid dispersion to enhance the dissolution rate of poorly water soluble Nifedipine [15].
Synthetic biodegradable polymer has been studied for its various applications mainly in controlled release system for drugs and biological [16-21]. Range of acrylic resins like Eudragit® RS, RL, E, L and S are commercially available and are widely used as coating materials in pharmaceutical industry; Eudragit® RL is the synthetic product of methacrylic acid ester with low content of quaternary ammonium group. They are insoluble in water and digestive juice, but swell and are permeable, which means that a drug can be release by diffusion [22]. Ethocel (ethyl cellulose) is hydrophobic polymer and used extensively as coating material, tablet binder and in preparation of microcapsules and micropsheres [23,24]. Gelatin is a water soluble polymer and due to its biocompatibility and biodegradability, is in fact an optimal candidate for producing controlled release systems for a wide variety of drugs [25,26].
The aim of this study is to improve the solubility of the poorly water soluble drug Felodipine by preparing solid dispersions using various hydrophilic carriers and using optimized solid dispersion to fabricate microcapsules for sustained release delivery of the drug with better bioavailability.
Materials and Methods
Materials
Felodipine was obtained from Cipla Ltd. Mumbai, India. Hydroxy propyl methyl cellulose (HPMC) was obtained as a gift from Blessings Pharmaceuticals Nagpur, India. Eudragit® RL 100 was obtained as a gift from Rohm Pharma GMBH, Germany. Gelatin was obtained from Shaw Wallace, India. Polyvinyl pyrrolidone was purchased from Loba Chemicals, India. Other reagents/chemicals were of AR/GR grade and purchased locally.
Methods
Preparation of Felodipine: HPMC, Felodipine and PVP solid dispersions: Solid dispersions of Felodipine: HPMC, Felodipine: PVP in different ratios were prepared by the solvent evaporation method. The formulations details of Felodipine: HPMC, Felodipine: PVP for solid dispersion are given in Tables 1 and 2 respectively. The specified amount of HPMC/PVP was weighed accurately and dissolved in minimum amount of hydro-alcoholic solution (1:1), Felodipine was added slowly with continuous kneading for 30 min. After sufficient kneading, the solvent was then allowed to evaporate. The dried mass obtained was then pulverized and sieved through mesh No. 200 (75 μ). Physical mixture was prepared by taking specified amount of HPMC / PVP and Felodipine in the glass mortar and it was triturated and mixed thoroughly. Then it was passed through mesh No. 200 (75 μ).
In-vitro release studies
In-vitro drug release from solid dispersions of Felodipine was tested using USP XXII Dissolution apparatus. The dissolution medium used was distilled water and bath temperature was maintained at 37 ± 1°C with paddle rotation speed of 100 rpm. Samples from the dissolution apparatus were withdrawn at 15 min time intervals and were analyzed for the % drug dissolved.
Fourier transforms infrared spectroscopy (FT-IR)
FT-IR spectroscopy was used to detect the existence of interactions between Felodipine and hydrophilic carriers. The FT-IR spectra of Felodipine, solid dispersion of Felodipine and HPMC were recorded in a potassium bromide pellet using Nicolet Magna FT-IR 550 series-II spectrophotometer.
Powder X-ray Diffraction (PXRD)
Powder XRD patterns of Felodipine, solid dispersion of Felodipine and HPMC were obtained to study the crystalline behavior of Felodipine, using Phillips XRD (model: 1700) with CμKα radiation (0.154 nm) at 35 kV and 20 mA over 2θ range.
Fabrication of microcapsules
Emulsion solvent evaporation technique [27] was employed to fabricate the Felodipine solid dispersion microcapsules using Ethyl cellulose or Eudragit® RL 100. Ethyl cellulose 0.5 g / Eudragit® 1 g was dissolved in 20 ml of acetone and solid Felodipine 2 g was suspended in it. This was emulsified into 200 ml of liquid paraffin, at 800 rpm using 1% Tween 80 as emulsifying agent. Phase separation and consequent coating of the core was achieved by solvent evaporation at room temperature. The microcapsules were filtered, washed with petroleum ether (60° to 80°C) to make them free of liquid paraffin and air-dried.
Coacervation-phase separation technique was employed to fabricate the Felodipine solid dispersion microcapsules using gelatin. Gelatin was dissolved in 10 ml distilled water, previously heated to 50°C, Felodipine was dispersed with continuous stirring, this drug suspension was then added drop wise to 100 ml of previously heated liquid paraffin containing 0.5 ml span 20. The mixture was stirred for 5 minutes to disperse the gelatin drug suspension as tiny droplet in oil phase. The temperature of entire system was then lowered down to 10°C. After stirring the mixture for 1 hr at 10°C, glutaraldehyde was added in different concentrations like 0.5 ml, 1.0 ml and 1.5 ml to different batches and stirring was continued for one and half hour. The beaker was covered and placed in freezer at -10°C for 24 hours for rigidization. After 24 hours, chilled n-hexane was added to slurry. The microcapsules were found to settle at the bottom of beaker, which was then separated by filtration. Then the microspheres were washed with petroleum ether (chilled) and were allowed to air dry for 24 hours.
Determination of In-vitro drug release
In vitro release of Felodipine from Felodipine solid dispersion microcapsules was evaluated using USP XXIII rotating paddle method. 50 mg microcapsules were tied in muslin cloth (mesh # 400) attached to the rotating paddle set at 100 rpm. Temperature of dissolution medium was 37 ± 0.1°C. 0.1 N HCl (pH 1.2) was used as a dissolution medium for first 2 h and phosphate buffer (pH 6.8) for 8 h. Samples were withdrawn at a specified time intervals, filtered and analyzed spectrophotometrically at 360nm. To maintain a constant volume, an amount of dissolution media equal to the volume of sample withdraw, was added immediately after each sample withdraw.
Scanning electron microscopy
The dried microcapsules were mounted on stubs with double adhesive tape and gold coated with sputter coater (Jeol J X A – 840 A, London, U.K.) for 120 seconds. The surface morphology of microcapsules was observed under a scanning electron microscope. (Stereo scans 250 MK-III, Cambridge England).
Encapsulation efficiency
Sieved microcapsules (50 mg) were grounded in a mortar and the drug content was extracted in pH 7.4 phosphates buffer. After suitable dilution of the sample, the drug content was analyzed spectrophotometrically at a wavelength of 360 nm. Every batch of microcapsule was analyzed in a triplicate.
Results and Discussion
Preparation of solid dispersion
Solid dispersion (SD) of Felodipine with PVP and HPMC were prepared with a view to improve its dissolution rate. All SD prepared were found to be fine and free flowing and gave fast and rapid dissolution of Felodipine when compared to Felodipine pure drug and physical mixture. Among the SD prepared with HPMC, H2 i.e. Felodipine: HPMC (2: 1) gave highest dissolution as shown in Figure 1. When D60 (time required to dissolve 60% of the drug) and D80 (time required to dissolve 80% of the drug) of H1, H2, H3 were compared, H2 showed lowest D60 and D80 values than H1 and H3. This indicates that less time was required by H2 to dissolve 60% or 80% of drug as compared to H1 and H3. When rate constant K (mg/min) of three SD and its physical mixture and pure drug was compared a 17-fold increase in dissolution rate of Felodipine was observed as shown in Table 1.
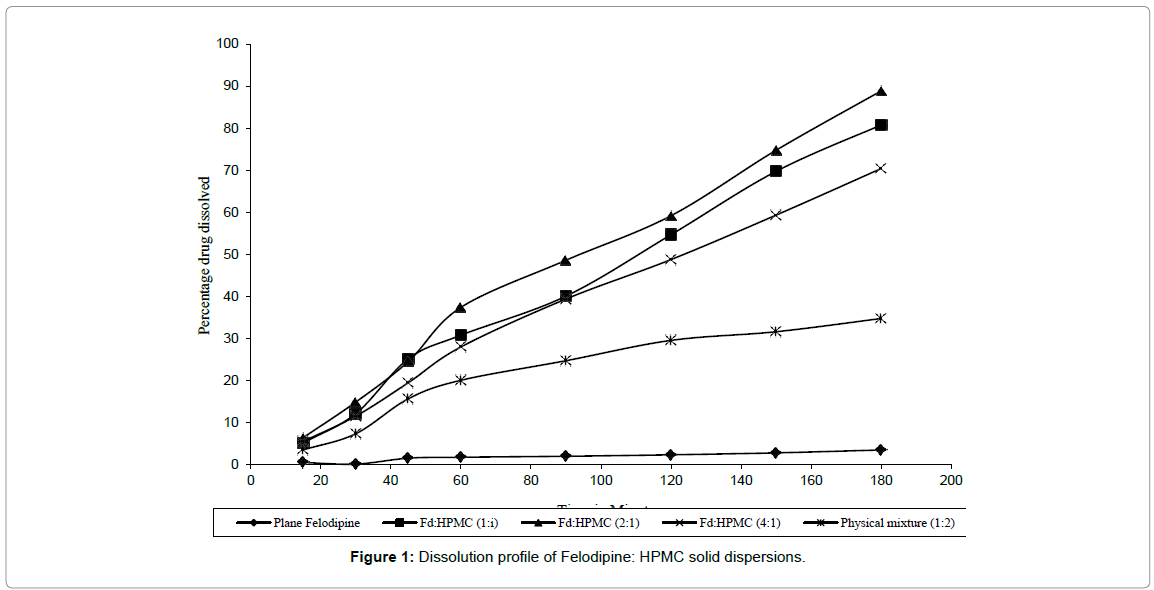
Figure 1: Dissolution profile of Felodipine: HPMC solid dispersions.
| Sr. No. |
Formulation Code |
Drug: Polymer |
D60 |
D80 |
K (mg/min) |
| 1. |
H1 |
1: 1 |
131 |
178 |
0.22 |
| 2. |
H2 |
2: 1 |
121 |
161 |
0.33 |
| 3. |
H3 |
4: 1 |
152 |
-- |
0.31 |
| 4. |
Physical mixture |
2: 1 |
|
|
0.12 |
| 5. |
Pure Felodipine |
|
|
|
0.019 |
Table 1: Details of solid dispersion of Felodipine using HPMC.
SD of Felodipine with PVP also showed increase in dissolution rate as compared to pure Felodipine as shown in Figure 2. When their D60 values were compared, SD P2 showed lower D60 value than P1 and P3 and their rate constants K (mg/min) also showed that SD P2 had higher drug dissolution rate than its physical mixture and SD P1 and P3. SD P2 showed 15 fold increases in dissolution as shown in Table 2.
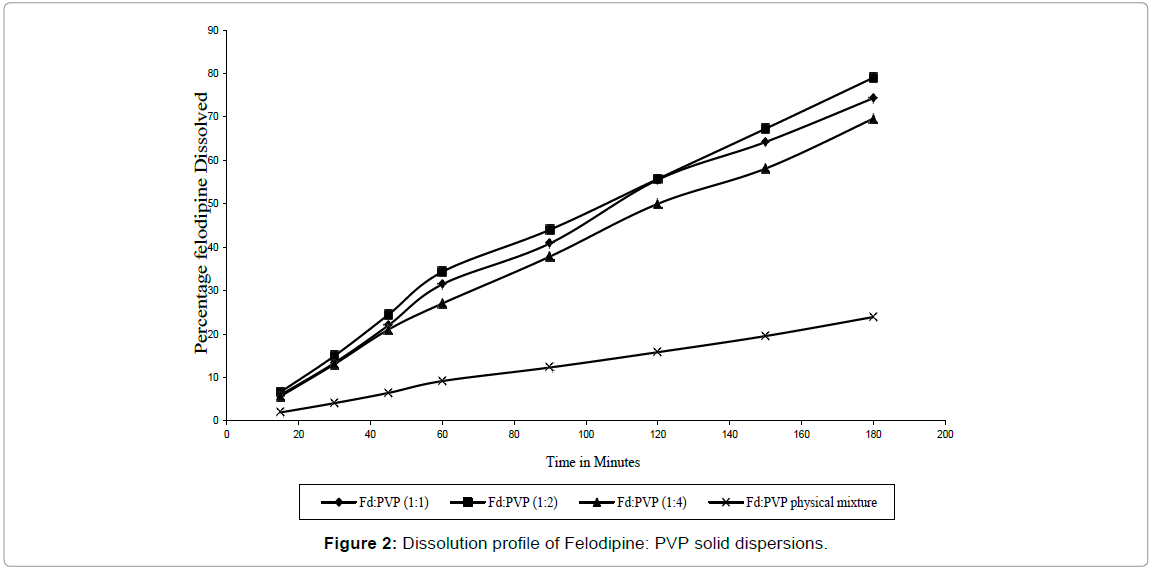
Figure 2: Dissolution profile of Felodipine: PVP solid dispersions.
| S.No. |
Formulation Code |
Drug : Polymer |
D60 |
D80 |
K (mg/min) |
| 1. |
P1 |
1 : 1 |
136 |
-- |
0.20 |
| 2. |
P2 |
2 : 1 |
121 |
182 |
0.28 |
| 3. |
P3 |
4 : 1 |
155 |
-- |
0.30 |
| 4. |
Physical mixture |
2 : 1 |
-- |
-- |
0.086 |
| 5. |
Pure Felodipine |
|
|
|
0.019 |
Table 2: Details of solid dispersion of Felodipine using PVP.
From the D60 and D80 and rate constant values of HPMC and PVP solid dispersion, it was observed that, HPMC improved the dissolution of Felodipine more effectively than PVP. Among the HPMC solid dispersion prepared in various ratios SD H2 (2:1) showed highest dissolution rate and was chosen for further physical characterization studies.
FT-IR Spectroscopy
One of the major problem concerning with the use of solid dispersion is their stability. If the molecular interaction between the drug and the excipients are not strong enough to keep the drug in an amorphous state or in a metastable crystalline form, the drug tends to transform back to stable crystalline form [28] these changes affect the chemical and physical properties of solid dispersion e.g. dissolution rate of drug. Therefore the solid state of Felodipine in solid dispersion and the type of interaction that occurs between Felodipine and the excipient were studied. FT-IR spectroscopy was used to detect the existence of interaction between Felodipine and hydrophilic carriers used during preparation of SD. When hydrogen bonding occurs between Felodipine and the carriers, a shift in certain-peaks, which OH affected by an interaction, can be observed in Felodipine spectra. In Felodipine, the groups in which hydrogen bonding can occur are the amine group in the ring and the two carbonyl group. When this hydrogen bonding occurs, bond energy at the N-H or C=O bond decrease and peak shift to lower frequencies is observed. This peak shift was most noticeable at the N-H stretch peak at 3369. 33 cm–1 and the C=O stretch peak at 1694.54 cm–1. But in the FT-IR spectra of Felodipine: HPMC it was observed that is no any shifting of N-H stretch peak and the C=O stretch peak, indicate the dispersion is devoid of any kind of interaction between the Felodipine and HPMC as carrier (Figure 3).
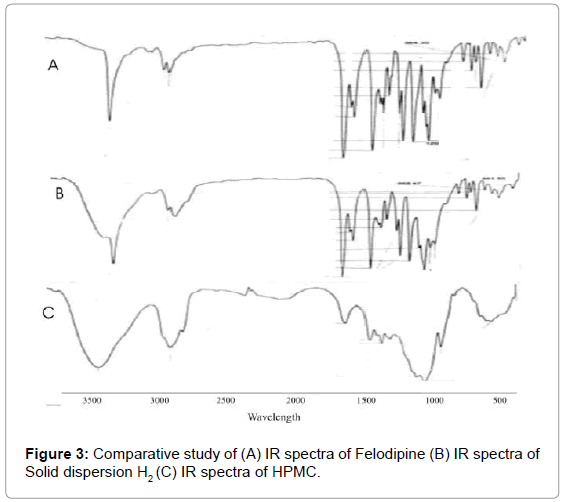
Figure 3: Comparative study of (A) IR spectra of Felodipine (B) IR spectra of Solid dispersion H2 (C) IR spectra of HPMC.
Powder XRD
XRD patterns of Felodipine: HPMC solid dispersions indicated changes in its thermodynamic and crystalline behavior (Figures 4 and 5) compared to thermo grams of pure Felodipine and physical mixture. The characteristics Felodipine peaks were found to reduce with Felodipine SD. These results provide evidence of decreased drug crystallinity due to the formation of a SD with HPMC - Felodipine.
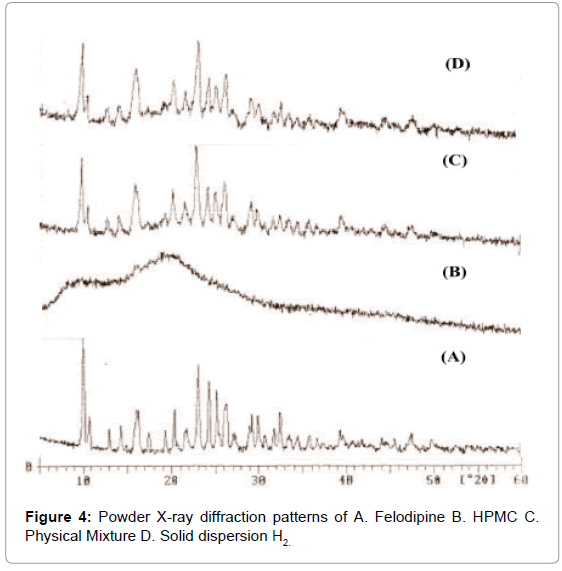
Figure 4: Powder X-ray diffraction patterns of A. Felodipine B. HPMC C. Physical Mixture D. Solid dispersion H2.
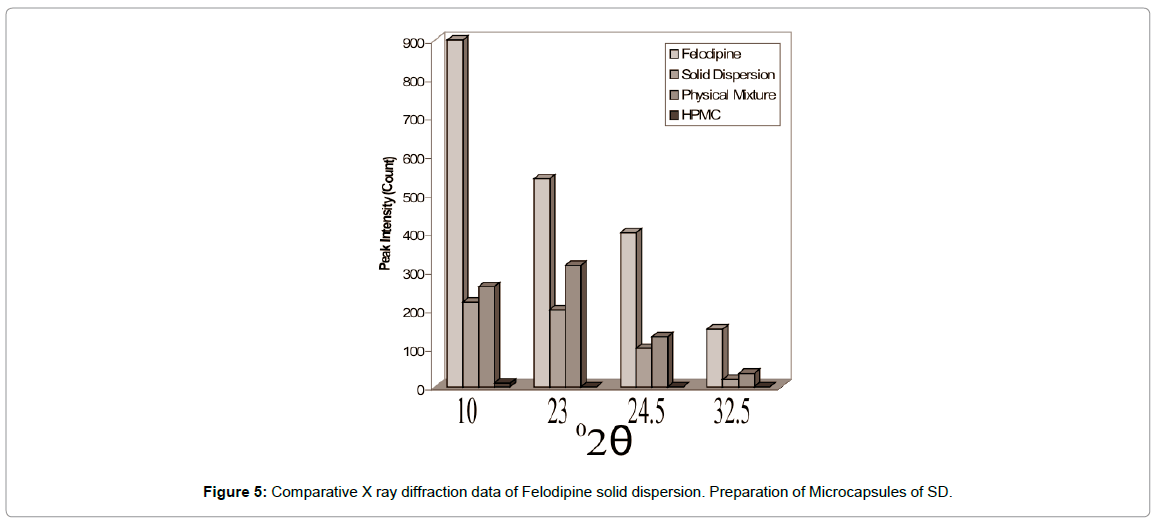
Figure 5: Comparative X ray diffraction data of Felodipine solid dispersion. Preparation of Microcapsules of SD.
A range of coating polymer from hydrophobic to hydrophilic nature were tried to prepare the microcapsules. Among the SD prepared with HPMC and PVP, the SD H2 with HPMC in the ratio (2: 1) gave fastest dissolution of Felodipine. Therefore, this SD was selected as core material for microencapsulation studies.
Firstly, ethyl cellulose, which is water insoluble, was tried as a coating polymer. The method used for microencapsulation was emulsion solvent evaporation method. The microcapsules obtained by this method were discrete, spherical and free flowing and the % entrapment efficiency ranging from (88-90%) as shown in Table 3.
| S. No. |
Formulation Code |
Core: Coat Ratio |
% Entrapment efficiency |
K (mg/min) |
| 1. |
EC1 |
4 : 1 |
88.09 |
0.12 |
| 2. |
EC2 |
6 : 1 |
89.99 |
0.025 |
| 3. |
EC3 |
9 : 1 |
90.4 |
0.054 |
Table 3: Effect of core: coat ratio on ethyl cellulose microcapsule properties (n=4).
From in vitro dissolution studies of ethyl cellulose microcapsule, the release rate constant were found to increase with increase in core: coat ratio from 4: 1 to 9: 1 with K (mg/min) 0.012 to 0.054 respectively. Whereas the dissolution rate constant K1 (mg/min) for SD H2 was found to be 0.33. In case of ethyl cellulose the release of drug was found to be diffusion controlled as the graph of amount of drug release Vs square root of time was found to be linear (Figure 6). Ethyl cellulose microcapsules with core: coat ratio 9: 1 release only 48.5% drugs in 8 hrs which was not satisfactory. Hence Eudragit® RL-100 was used which is freely permeable to water for better and quicker release of drug.
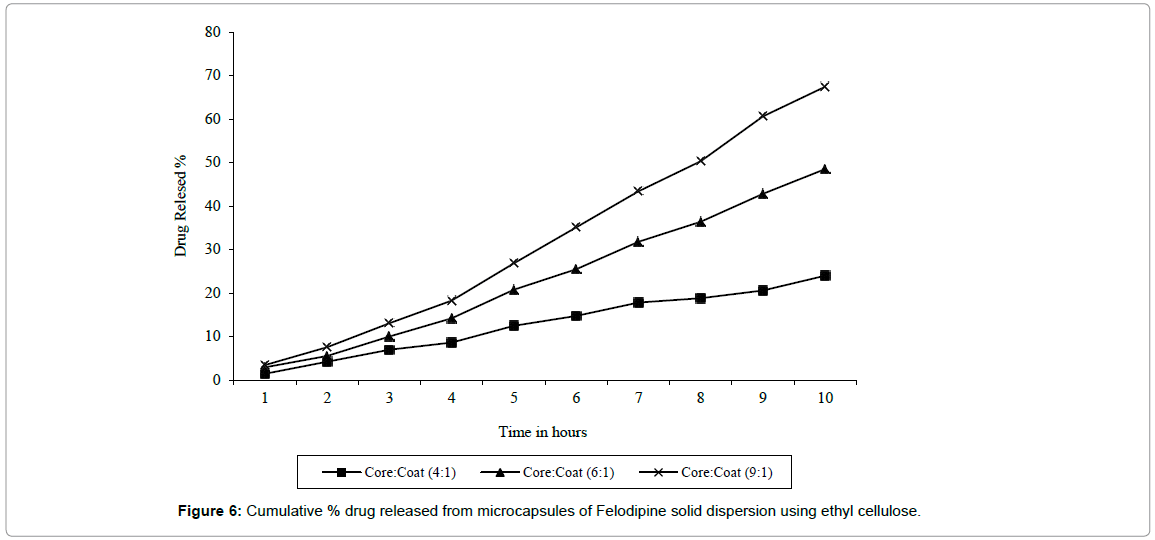
Figure 6: Cumulative % drug released from microcapsules of Felodipine solid dispersion using ethyl cellulose.
Microcapsules prepared by using Eudragit® RL-100 were found to discrete, spherical and free flowing. The % entrapment efficiency was found to be good (85-92%). The release rate constant for various batches of microcapsules in the core: coat ratio 2: 1, 4: 1, 6: 1 and 9:1 was found to be 0.0075, 0.020, 0.038, and 0.072 respectively as given in Table 4. The release of drug from Eudragit® RL-100 microcapsules may be due to permeation of aqueous fluid through the polymeric membrane and leaching of the drug out of the coating polymer [22]. The comparative dissolution profile of drug release from different batches of microcapsules prepared with different core–coat ratios are shown in Figure 7 with an increase in the proportional concentration of core to coat, the faster the release rate of drug from microcapsules. This may be attributed to the reduced wall thickness of the microcapsules. The present results are in agreement with previous studies [29] that increasing the core: wall ratio results in an increase in the dissolution rate due to a decrease in coat thickness ultimately reducing the distance traveled by the drug through/within the coat. Microcapsules with highest core: coat ratio i.e. 9: 1 showed 67.44% drug release in 8 hrs. Hence a complete hydrophilic polymer i.e. gelatin was selected for microencapsulation of Felodipine solid dispersion to get proper [30,31].
| S. No. |
Formulation Code |
Core: Coat Ratio |
% Entrapment efficiency |
K (mg/min) |
| 1. |
EDRL-1 |
2 : 1 |
85.27 |
0.0075 |
| 2. |
EDRL-2 |
4 : 1 |
88.24 |
0.020 |
| 3. |
EDRL-3 |
6 : 1 |
90.12 |
0.038 |
| 4. |
EDRL-4 |
9 : 1 |
92.67 |
0.072 |
Table 4: Effect of core: coat ratio on Eudragit® microcapsule properties (n=4).
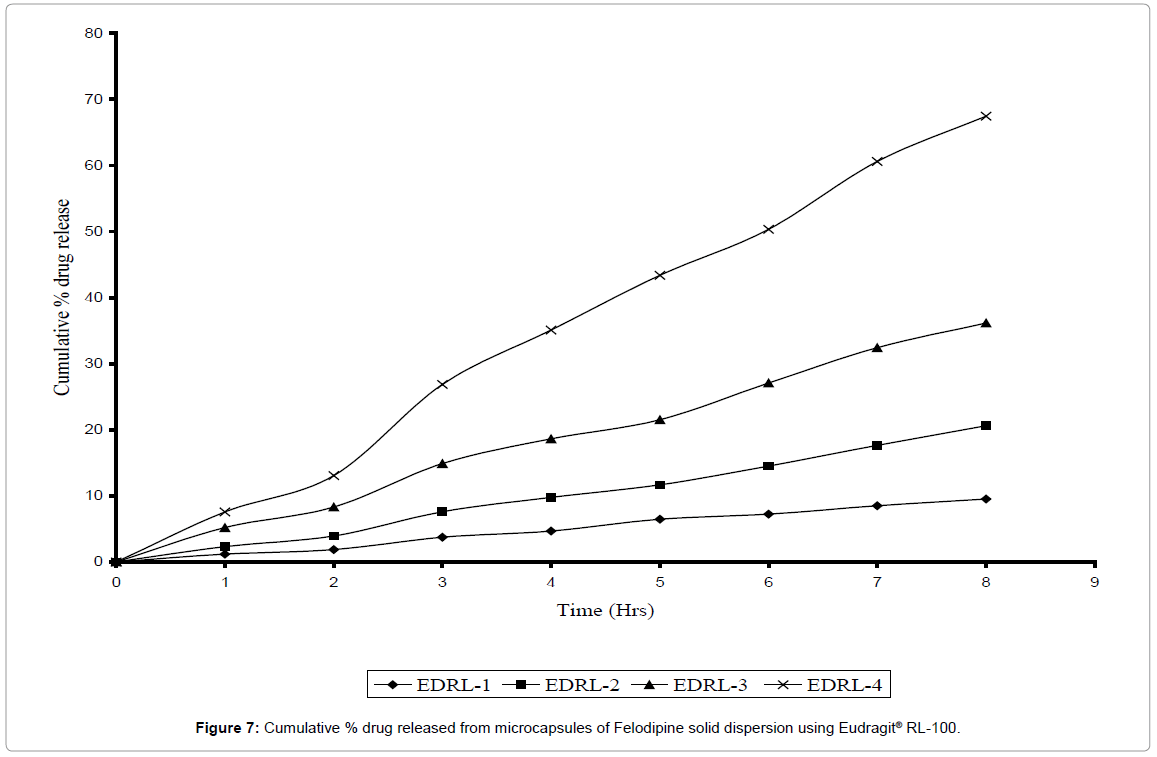
Figure 7: Cumulative % drug released from microcapsules of Felodipine solid dispersion using Eudragit® RL-100.
Microcapsules prepared with gelatin as wall forming agent were also found to be spherical and free flowing. The % entrapment efficiency was found to be within 88-96% range. Microcapsules were prepared using two core: coat ratios i.e. 1: 1 and 2: 1 and the varied amount of glutaraldehyde (0.5 ml, 1.0 ml, 1.5 ml) was added as a cross linking agent. The cross linking time was kept constant for all batches. In vitro dissolution studies of six different batches of gelatin microcapsule were carried out and their release rate constant K and their D60 values were determined as shown in Tables 5 and 6. Release rate was found to be dependent on the amount of glutaraldehyde added. Release rate constant was found to decreasing as the concentration of glutaraldehyde was increased where as their D60 values were found to increase with increase in concentration of glutaraldehyde. The Felodipine release from gelatin microcapsules was 72% and 86% in different core: coat ration as 1:1 and 1:2 respectively (Figures 8 and 9) over the period of 8 hrs. The G2 – I batch with core: coat ratio 2: 1 with 0.5 ml glutaraldehyde gave slow, controlled and complete release spread over period of 8 hrs. With highest release constant of 0.077 and slowest D60 values of 5-6 hrs. The rate constant of various microcapsules of 3 different polymers was found to decrease in the order Gelatin > Eudragit® RL-100 > Ethyl cellulose.
| S.No. |
Formulation Code |
Amount of Glutaraldehyde |
% Entrapment efficiency |
K (mg/min) |
| 1. |
G1 – 1 |
0.5 |
92.98 |
0.046 |
| 2. |
G1 – 2 |
1.0 |
90.15 |
0.039 |
| 3. |
G1 – 3 |
1.5 |
87.55 |
0.030 |
Table 5: Effect of glutaraldehyde on microencapsulation of Felodipine solid dispersion using gelatin (core: coat 1:1).
| S.No. |
Formulation Code |
Amount of Glutaraldehyde |
% Entrapment efficiency |
K (mg/min) |
| 1. |
G2 – 1 |
0.5 |
96.26 |
0.077 |
| 2. |
G2 – 2 |
1.0 |
95.18 |
0.065 |
| 3. |
G2 – 3 |
1.5 |
88.27 |
0.049 |
Table 6: Effect of glutaraldehyde on microencapsulation of Felodipine solid dispersion using gelatin (core: coat 2:1).
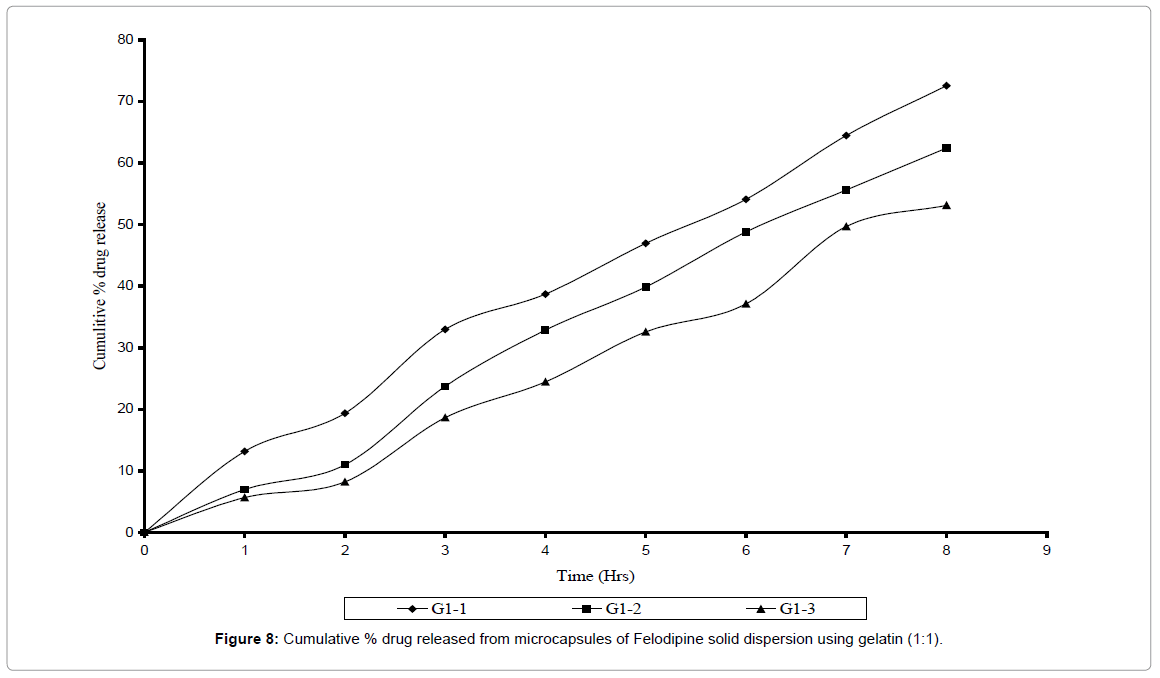
Figure 8: Cumulative % drug released from microcapsules of Felodipine solid dispersion using gelatin (1:1).
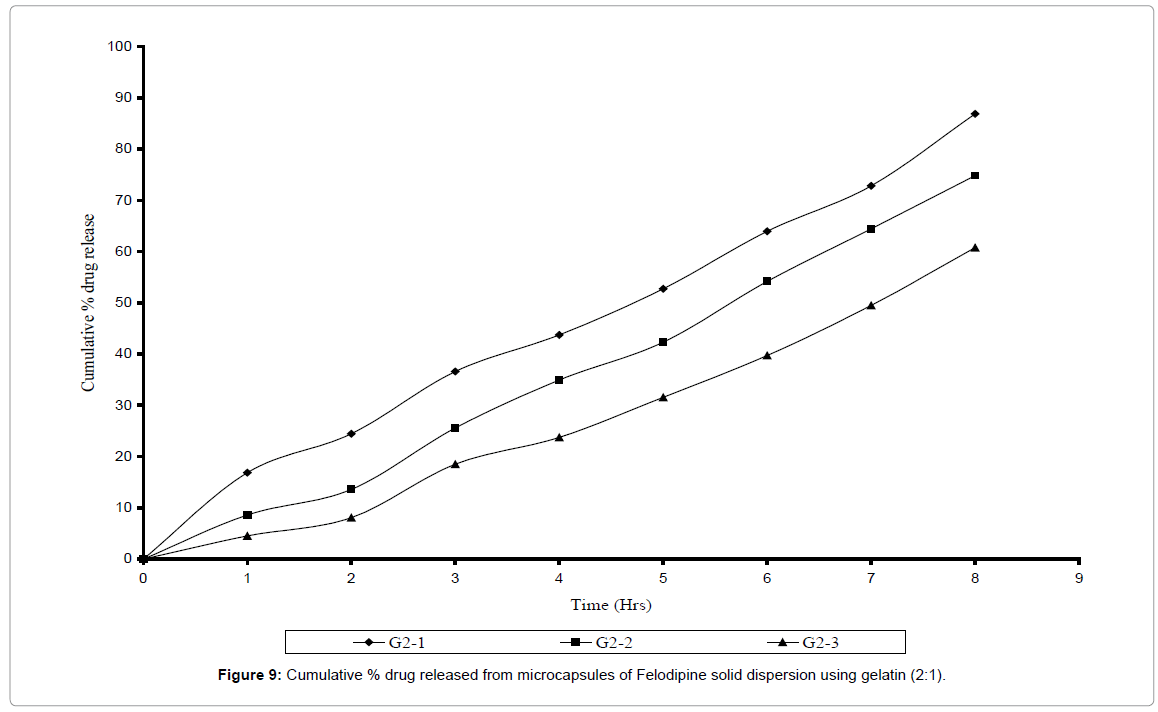
Figure 9: Cumulative % drug released from microcapsules of Felodipine solid dispersion using gelatin (2:1).
Characterization of microcapsules
As satisfactory results were obtained with gelatin microcapsules (G2-I), this batch was used for further characterization studies. IR studies were carried out to study any physicochemical interaction between coating polymer and the core material. The IR spectrum of gelatin microcapsule shown in Figure 10 was similar to that of pure Felodipine and it’s SD. The principle IR absorption peaks of Felodipine at 1205 cm–1 (-C-O ether), 1381 cm–1 (-C-H-), 1620 cm–1 (-C=Caromatic), 1694 cm–1 (-C=O ester) were observed along with 669 cm–1 [-C-H alkenes ( C =C) ] of gelatin. These spectral observations indicates that there was no physicochemical interaction between core materials as both drug and excipient were present in its original form without any degradation.
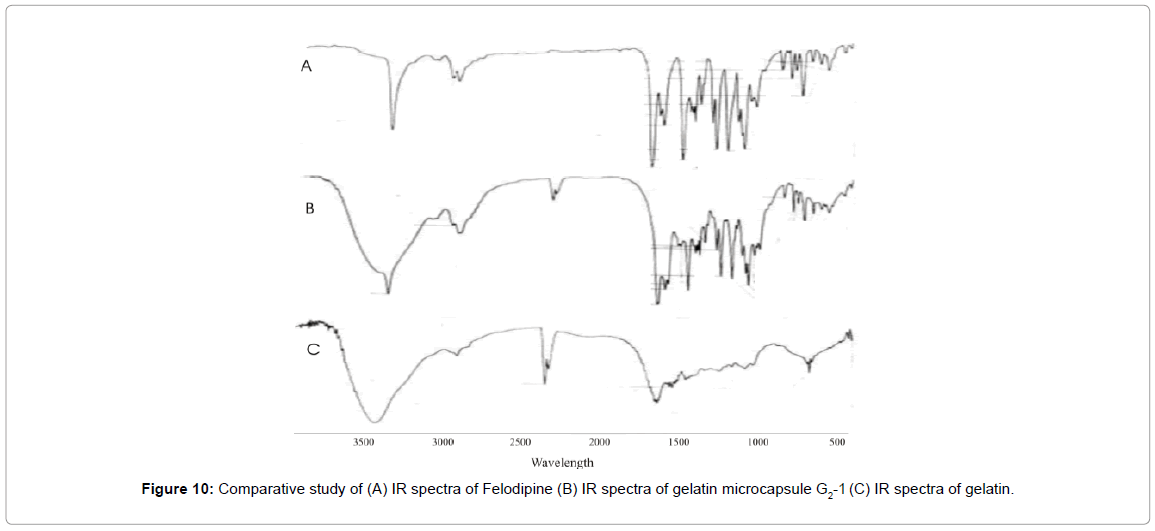
Figure 10: Comparative study of (A) IR spectra of Felodipine (B) IR spectra of gelatin microcapsule G2-1 (C) IR spectra of gelatin.
XRD pattern was recorded to study the physical nature of the core material. By using the technique of SD highly crystalline pure Felodipine was converted to its amorphous form in its SD, which was confirmed by XRD patterns of SD. SD of Felodipine was microencapsulated to formulate extended release dosage form using gelatin. The XRD pattern of microcapsule was recorded to study the polymorphic changes that occur in core material during fabrication of microcapsules.
The XRD pattern of gelatin microcapsules as shown in Figures 11 and 12 shows comparison of XRD data of Felodipine, solid dispersion and gelatin microcapsules and reveal that these was still considerable reduction in the intensity of four major diffraction peaks at 10, 23, 24.5 and 32.5 of pure Felodipine as similar to that of solid dispersion showing the core material was still in its amorphous phase and no polymorphic changes occurred during microencapsulation process.
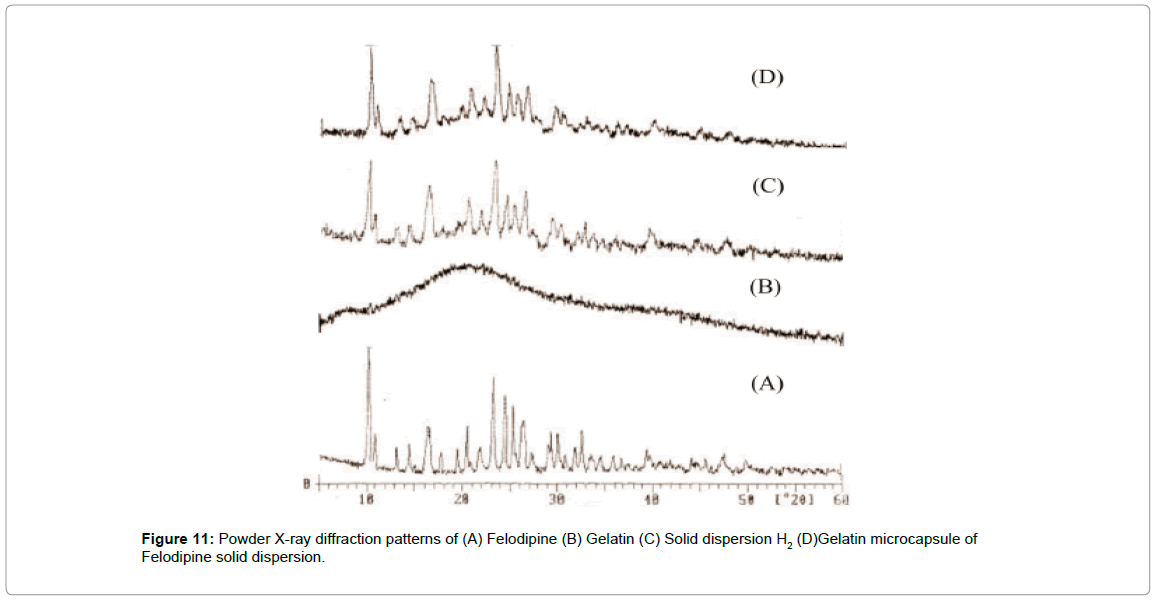
Figure 11: Powder X-ray diffraction patterns of (A) Felodipine (B) Gelatin (C) Solid dispersion H2 (D)Gelatin microcapsule of Felodipine solid dispersion.
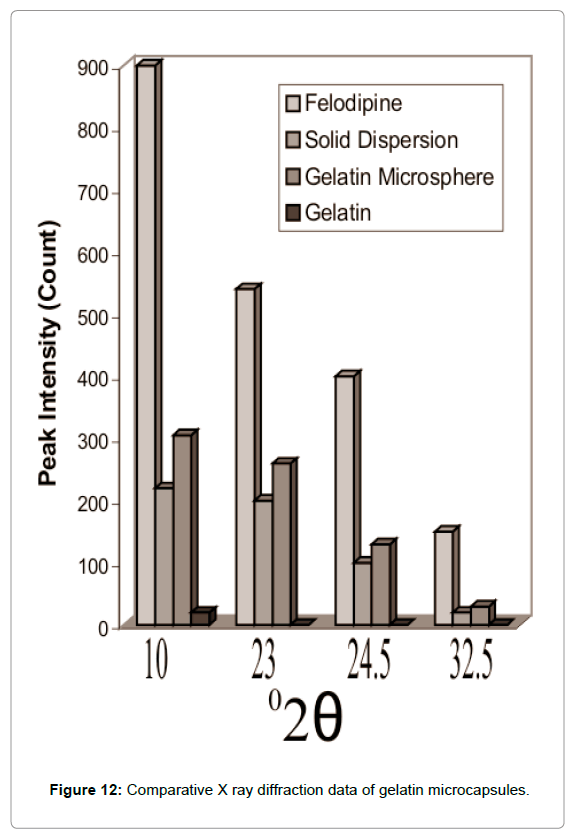
Figure 12: Comparative X ray diffraction data of gelatin microcapsules.
The microcapsules of three different polymers were characterized by SEM studies which were carried out at different magnification (Figures 13-15) to study the shape and surface morphology of microcapsule. All microcapsules were discrete spherical and were found to be covered with continuous coating of polymer.
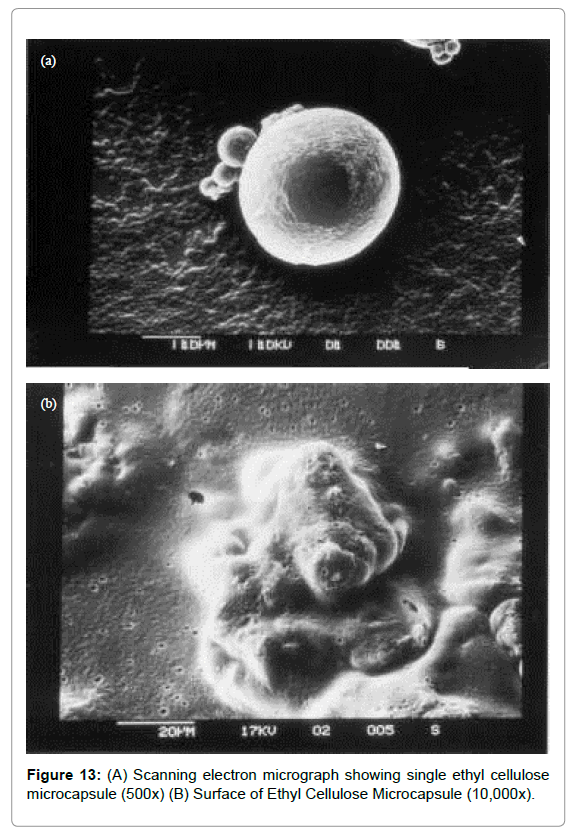
Figure 13: (A) Scanning electron micrograph showing single ethyl cellulose microcapsule (500x) (B) Surface of Ethyl Cellulose Microcapsule (10,000x).

Figure 14: Scanning electron micrograph showing (A) Uniform Eudragit® RL-100 Microcapsule (500x) (B) Surface of Eudragit® RL-100 Microcapsule (10,000x).
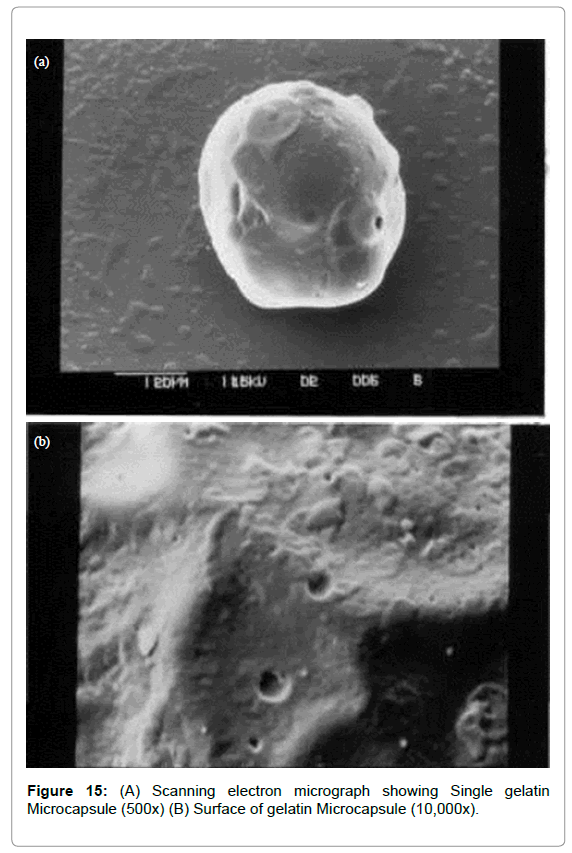
Figure 15: (A) Scanning electron micrograph showing Single gelatin Microcapsule (500x) (B) Surface of gelatin Microcapsule (10,000x).
Conclusion
Extended release dosage forms of Felodipine are difficult to formulate due to its poor solubility. Approach of solid dispersion was employed in which hydrophilic carriers like HPMC and PVP were used. HPMC markedly enhanced the dissolution rate of Felodipine. XRD studies clearly indicated that highly crystalline state of Felodipine is converted to amorphous form in solid dispersion. IR studies also indicated no chemical interaction between Felodipine and excipient. Thus, solid dispersion was then microencapsulated using different coating material using emulsion solvent evaporation and coacervation phase separation method. Microcapsules formulated were found to be spherical and discrete as seen in SEM photomicrographs and were also having good encapsulating efficiency. Felodipine- HPMC solid dispersion microencapsulated with gelatin (G2) gave better performance than micro capsulated with ethyl cellulose and Eudragit® RL. Felodipine release up to 86% after 8 hrs was observed. Core material was still existed in its amorphous form in microcapsules without any polymorphic changes which was confirmed by XRD. Drug release depended on the core: coat ratio and the amount of glutaraldehyde added.
Acknowledgement
Authors are thankful to Head, Department of Pharmaceutical Sciences, Nagpur University, Nagpur for providing laboratory facilities.
24589
References
- Ford JL (1986) The current status of solid dispersions. Pharmaceutica Acta Helvetiae 61: 69-88
- Lin SL, Menig J, Lachman L (1968) Interdependence of physiological surfactant and drug particles on dissolution behavior of water insoluble drugs. J Pharma Sci 57: 2143-2150.
- Kornblum SS, Hirschaorn JO (1970) Dissolution of poorly water-soluble drugs. J Pharm Sci 56 :606-614.
- Parrot EL (1974) Milling of pharmaceutical solids. J Pharma Sci 63: 813-820.
- Chiou WL, Riegelman S (1971) Pharmaceutical application of solid dispersion systems. J Pharma Sci. 60: 1281-1302.
- Serajuddin ATM (1999) Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems and recent breakthroughs. J Pharm Sci 88: 1058-1066.
- Sheen P, Khetarpal VK, Cariola CM, Rowlings CE (1995) Formulation studies of poorly water soluble drug in solid dispersion to improve bioavaibility. Int J Pharm 118: 221-227.
- Shah JC, Chen JR, Chow D (1995) Preformulation study of etoposide: II. Increased solubility and dissolution rate by solid-solid dispersions. Int J Pharma 113: 103-111.
- Mooter VG, Augustijns P, Blaton N, Kinget R (1998) Physico-chemical characterization of solid dispersion of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm. 164: 67-80.
- Tantishaiyakul V, Kaewnopparat N, Ingkataworn WS (1996) Properties of solid dispersion of piroxicam in polyvinylpyrrolidone K-30. Int J Pharm 143: 59-66.
- Torrado S, Torrado JJ, Cadorniga R (1996) Preparation dissolution and characterization of albendazole solid dispersion. Int J Pharm 140: 247-250.
- Ho H, Su H, Tsai T, Sheu M (1996) The preparation and characterization of solid dispersion on pellets using a fluidized bed system. Int J Pharm 139, 223-229.
- Perng CY, Kearney AS, Patel K, Palepu NR, Zuber G, et al. (1998) Investigation of formulation approaches to improve the dissolution of SB-210661, a poorly water soluble 5-lipoxygenase inhibitor. Int J Pharm 176: 31-38.
- Nair R, Gonen S, Hoag SW (2002) Influence of polyethylene glycol and povidone and the polymorphic transformation and solubility of carbamazepine. Int J Pharm 240: 11-16.
- Mehta KA, Kislalioghu MS, Phuapradit W, Malick W, Shah NH (2002) Multi unit controlled release systems of nifedipine and nifedipine: Pluronic F68 solid dispersions: Characterization of release mechanism. Drug Dev Ind Pharm 28: 275-286.
- Langer RS, Peppas NA (1981) Present and future applications of biomaterials in controlled drug delivery systems. Biomater 2: 201-214.
- Heller J (1984) Zero order drug release from bioerodible polymer: In Advances in Drug Delivery Systems, Plenum Press: New York, USA. pp. 101-121.
- Tice TR, Cowsar DR (1984) Biodegradable controlled release parenteral systems. Pharm Tech 8: 26-35.
- Kitchell JP, Wise DL (1985) Poly (Lactide/glycolide) biodegradable drug –polymer matrix system. In Methods in enzymology: Academic Press: Orlando FL, USA. 112: 436-448.
- Juni K, Nakano M (1987) Poly (hydroxyl acids) in drug delivery, Critical Review of Therapeutic Drug Carrier System Crit Rev Ther Drug 3: 209-232.
- Linhardt RJ (1989) Biodegradable polymers for the controlled release of drugs. In Controlled release of drugs: Polymers and aggregate system, VCG Publishers: New York, USA. pp. 53-95.
- Shigeru G, Masakazu K, Masahiro N, Kaori M, Toshinobu A, et al. (1986) Eudgragit RS and RL (acrylic resins) microcapsules a pH insensitive a sustained release preparation of ketoprofen. J. Microencapsulation 3: 293-304.
- Jalsenjak I, Nicolaidou CF, Nixon JR (1976) The in vitro dissolution of phenobarbitone sodium from ethyl cellulose microcapsules. J Pharm Pharmacol 28: 912-914.
- Tirkkonen S, Paronen P (1992) Enhancement of drug release from ethylcellulose microcapsules using solid sodium chloride in the wall. Int J Pharm 88: 39-51.
- Dupar KC (1987) Method of preparing gelatin microcapsule. Drug Dev Ind Pharm. 13: 1023-1030.
- Narayani R, Panduranga R (1993) Preparation, characterization and in vitro stability of hydrophilic gelatin microspheres using a gelatin-methotrexate conjugate. Int J Pharm 95: 11-15.
- Amperiadou A, Georgarakis M (1995) Controlled release salbutamol sulphate microcapsules prepared by emulsion solvent-evaporation technique and study on the release affected parameters. Int J Pharm 115: 1-8.
- Fromming KH, Hosemann R (1985) Stability problems under special consideration of solid dispersions of drugs. STP Pharma. 1: 660-665.
- Lavasanifar A, Ghalandari R, Ateai R, Zolfaghari ME, Mortazavi SA, et al. (1997) Microencapsulation of theophylline using ethyl cellulose: In vitro drug release and kinetic modelling. J. Microencapsulation 14: 91-100.
- Wood DA (1980) Biodegradable drug delivery systems. Int J Pharm 7: 1-18.
- Rao BC, Rao VS (1976) Study of microcapsules prepared by coacervation-phase separation, Eastern Pharmacist 19: 127-129.


