Keywords
Lassa virus, nucleoprotein, arena viruses,
Introduction
Lassa virus belongs to a very large group of heamorrhagic fever viruses-arena viruses which is responsible for high rate of morbidity and mortality in areas of West Africa. Among the six arena viruses known so far only one –Lassa virus (LASV) is known to cause illness in humans. There are an estimated 300,000 to 500,000 cases of Lassa fever each year [1,2,3,4,5] with mortality rate of 15% to 20% for hospitalized patients and as high as 50% during epidemics [2,6,7]. The LASV genome is comprised of two ambisense, single stranded RNA molecules, designated small (S) and large (L) [8]. Two genes on the S segment encode NP, GP1, and GP2; whereas, the L segment encodes the viral polymerase (L protein) and RING finger Z matrix protein. GP1 and GP2 subunits result from post-translational cleavage of a precursor glycoprotein (GPC) by the protease SKI-1/S1P [9,10]. GP1 serves a putative role in receptor binding, while the structure of GP2 is consistent with viral transmembrane fusion proteins [11,12].
Over the years many aspects of Lassa fever have been defined including the clinical presentation, epidemiology, immunology, pathology, physiology and therapy [13,14] with sparse reports on molecular evolution. The exact seasonality of Lassa virus is still not understood and therefore increase in availability of rodent host (Mastomys natalensis) may increase the risk of infection or transfer of the virus to humans. Lassa virus infection has continued unabated despite the presence of circulating antibodies against the infection in the areas of epidemics with emergence of newer strains which might likely be the cause of new outbreaks [15], hence a detailed evolutionary study at molecular level becomes essential. Serological data did not provide information on seasonality and the origin of human to human infection.
Lassa virus was first identified in Northern Nigeria in 1969, although reports have it that the virus co-evolved with the rodent host over a period of 9 million years. A study of the molecular evolution will provide better and concise information as to the origin of the virus in humans and its seasonality. The recent identification of two new strains point to the fact that the virus is in constant genetic change which has made this study necessary. Evolutionary study of the virus which is a determinant of the virulence diversity have been reported to give a knowledge of possible vaccine development for the virus and also give an in site into the host parasite relationship. A further study may eventually lead to the explanation as to why despite the circulating antibodies against Lassa virus, outbreaks still occur yearly.
Methods
The computer program MEGA 4 and CLC workbench 4 enabled us to separately estimate the rate of synonymous and nonsynonymous substitution as wells as study the entire evolution of Lassa virus nucleoprotein using 18 nucleotide sequences identified over the years and deposited in Genbank which included their genetic distances, evolutionary rate, neighbor joining methods of evolutionary studies, phylogenetic tree, substitution frequency and pattern. The genetic distances between strains were estimated using pairwise nucleotide differences. The substitution pattern as well as transition and tranversional substitution and frequency of parallel independently occurring at the same site of different evolutionary lineages were studied. Multiple sequence alignment of the sequences were also carried out and viewed in genedoc to observe for evolutionary conservation.
Nucleotide Sequences
Sequences used in this study were from Genbank and are as follows:
L._virus_DGD112_NP, L._virus_DGD104_NP, L._virus_DGD87_NP,
L._virus_DGD43_NP, L._virus_DGD35_NP, L._virus_DGD28_NP,
L._virus_DGD13_NP, L._virus_DGD4_NP, L._virus_TA846_NP,
L._virus_TA820_NP, L._virus_TA817_NP, L._virus_TA491_NP,
L._virus_TA471_NP, L._virus_TA464_NP, L._virus_TA462_NP,
L._virus_TA444_NP, L._virus_TA341_NP, L._virus_TA416_NP.
A graph of evolutionary differences representing their evolutionary distance was plotted against the time of identification of the new strain in order to determine the time of emergence of the virus using only human isolates since 1972. The dates of isolation of all the human strains used were extracted from Bowen et al. [16].
Results
Alignment of the 18 nucleotide sequences of Lassa virus strains demonstrated insertion and deletions as well as substitutions among the strains collected over the years from different parts of the world. It also showed some conserved areas over the years especially regions of suspected high functionality. Of the 628 sites of substitution, 488 were conserved. There were 399 nondegenerate sites, 101 two fold degenerate sites and 46 four fold degenerate sites.
The rate of synonymous substitution was high 5.889 per nucleotide per year and nonsynonymous was higher at 49.664. The average predicted rate of synonymous and nonsynonymous using modified Nei-Gojobori (assuming transition/transversion bias = 2) was 27.9 which was taken as the genetic distance between strains. The average number of synonymous sites is 150.741. The average number of nonsynonymous sites is 392.259. The phylogenetic tree was inferred by unweighted pairwise grouping in MEGA4 and using neighbour-joining method (Fig. 1). The time of emergence of Lassa virus was predicted to be around January 1920 (±24 months). And the time of emergence of the virus in humans was predicted to be May 1959 about ten years before the first Lassa virus report in Nigeria. In synonymous substitution the rate of (G-----T) was high. The nucleotide frequencies were 0.314 (A), 0.246 (T/U), 0.204 (C) and 0.235 (G). The transition/transversion ratio k1=14.991 (purines) and k2=69.916 (pyrimidines). The overall transition/transversion bias R=16.662 with a total of 620 position in the final data set. These figures are far higher than an earlier study using lassa virus glycoprotein. The nucleotide diversity was also very high using the Taijima’s model in MEGA 4. Of the 628 sites 399 were non degenerate while 101 were two fold degenerate and 46 four fold degenerate. The neighbour joining method was used to explore the evolutionary relationship of isolates using Kt as the genetic distance (Fig. 1). This is based on the principle that the sum of the branch length at each cluster stage is to be minimised. Thus this method is suitable for estimating genetic relationship among isolates without taking into account evolutionary rate or isolation time. The nucleotide diversity was high at 0.099905. Amino acid substitution was high where 99 of the total 209 sites were conserved. The graph of the nucleotide distance against their time of identification of new human strains (Fig 2) predicted the first human infection to be around May 1959 using the 1972 Josiah strain as a reference strain.
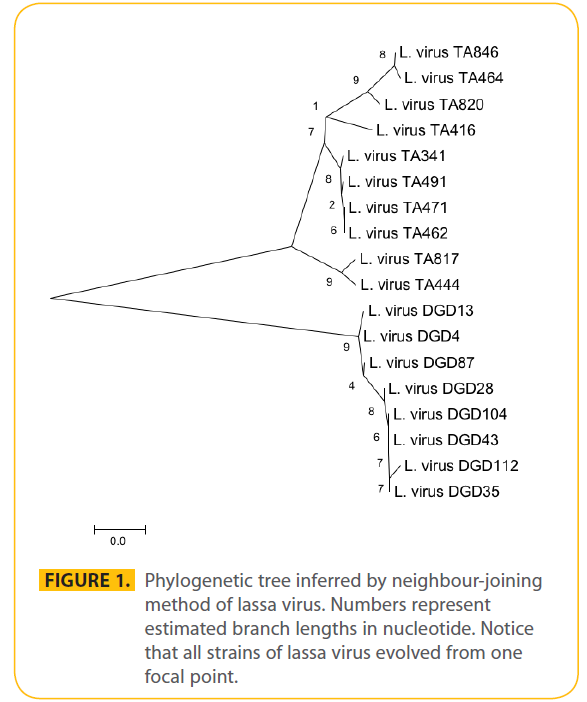
Figure 1: Phylogenetic tree inferred by neighbour-joining method of lassa virus. Numbers represent estimated branch lengths in nucleotide. Notice that all strains of lassa virus evolved from one focal point.
Figure 2: Evolution rate of Lassa virus and the first human infection. The 1972 strain was taken as the stand point for first case. Using 4 strains and assuming that they all evolved at a constant rate, the evolutionary distance was plotted against time of emergence. A dotted line from the point of joins at the dot plot, backwards will terminate at the month of first emergence. Or the slope of the plot in months minus the proposed first emergence date will give the date of first emergence of human straisn. The dot plot was done in SPSS version 17.
Discussion and Conclusion
The phylogenetic tree was inferred with 18 Lassa virus nucleoprotein nucleotide sequences using MEGA 4 and this was consistence with results from other studies [17,18]. Okoror et al., [18] studied the Lassa virus glycoprotein nucleotide sequences using MEGA 4 while Takeda compared 18 nucleotide of capsid protein VP1 in 18 isolates of EV70 in two separate studies with earlier study using oligonucleotide mapping of the entire genome [19]. The emergence of Lassa virus was predicted to be 1920 (±24 months) which opposes earlier studies that suggested that the virus probably co-evolve millions of years ago with the rodent host. However the rodent strain might be different from the human strain since all the strains compared in this study were all human strains. It is suggested that as soon as the strains crosses the species barrier to infect humans there will be a mutation in the nucleotide sequences. This study also revealed that the divergence times are usually during the period of high epidemics which confirms that the emergence of newer strains were responsible for the yearly epidemic outbreaks. The concentration of strains at the period of epidemics also inferred that the variations in strains is usually location dependent rather time dependent. The occurrence of epidemics at the same time in all the endemic areas also supportive of the fact that newer strains are responsible for epidemics in different locations. The fact that epidemics occur at the same time in different areas by different strains of the virus confirms earlier report by Banseh [20] who suggested that the virus vary with location. In this study therefore we suggest that the virus vary according to both location and time. The high rate of transition/transversion at the same time shows that there is co-circulation of genetically and antigenically different strains in the population. This goes to point that the nucleoprotein is suitable for evolutionary studies but the glycoprotein is more suited for vaccine development if the study by Okoror et al. [18] is anything to compare with.
The nucleotide substitution rates of the Lassa virus nucleoprotein gene obtained here (27.6) weighted average for both synonymous and nonsynonymous per nucleotide per year were higher than those of an earlier study by Okoror et al., [18]. This goes to confirm that the Lassa virus nucleoprotein is best suited for evolutionary studies than the glycoprotein since it gives a better insight of the periodic nucleotide changes especially with a nucleotide diversity as high as 0.099905. This is in opposition to earlier studies in non-structural protein gene [21], nucleoprotein gene [21,22] and PB2 of influenza [23] A virus and for haemagglutinin [24] and NS genes [25] of influenza B virus; they were in the range of 1.1 x 10-3 to 2.3x10-3 substitutions per nucleotide per year for the observed nucleotide differences. There was a high remarkable feature of the nucleotide substitution patterns of Lassa virus transitions, i.e., low transversions. The relative frequency of transitions (C*- -U and A*- ->G) was almost 70% (R = 16.662 in the 620 sites in the final data sets in all three estimations, i.e., in total substitutions, at the third position of codons, and at fourfold-degenerate sites.
This is an indication that transitions were found in almost all the sites in the data set. Although transitions in naturally occurring mutations are known to be much more frequent than expected from random substitutions [26,27], the frequency of transitions in Lassa virus glycoprotein gene was much higher than that reported for the pseudogene DNA of human origin (55.6% on average) [26], for the third codon position of the Mahoney and Sabin strains of type 1 poliovirus (77.1%). This study is in opposition with earlier report by Takeda et al. [17] which suggested that the short period for the analysis of the EV70 VP1 gene was responsible for its high transition since this study analysed sequences that were isolated close to 40 years ago. Such high frequency was also observed for Lassa virus glycoprotein which was analysed for a shorter period of time. Hence the time of isolation is not a parameter to judge is transition. This study also suggests that virulence diversity in arenaviruses as studied is essential for the development of vaccine against the virus as earlier reported by Jahrling and Peters and other reports [28,29,30]. It is also suggested that the reasons for a yearly epidemic outbreak is due to the fact that each time the rodents strain crosses the species barrier there is always a re-modification of genetic or molecular make-up of the virus, hence not been recognised by already circulating antibodies.
Author Contribution: OLE conceived the study and did all the computational study as well as wrote the paper. OOI was actively involved in interpretation of the results and the proof reading of the final manuscript up to the point of submission. She also supervised most of the evolutionary analysis.
Conflicting Interest: There was no conflicting interest.
2552
References
- McCormick JB (1986) Clinical, epidemiologic, and therapeutic aspects of Lassa fever. Med Microbiol Immunol 175; 153-155.
- McCormick JB (1987) Epidemiology and control of Lassa fever. Curr Top Microbiol Immunol, 134; 69-78.
- Andrea G (1993) Molecular approach for treatment for treatment of haemorrhagic fever virus infection. Antiviral Research 22 (1) 45-75.
- Agbonlahor DE (1998) Inaugural lecture: mankind exploited, microbes revolted and their maker intervened 23-27.
- Fisher-Hoch SP, McCormick JB (2004) Lassa fever vaccine: A review. Expert Rev Vaccines 3(2) 103-111.
- Fisher-Hoch SP, Tomori O, Nasidi A, Perez-Oronoz GI, Fakile Y, Hutwagner L, McCormick JB (1995) Review of cases of nosocomial Lassa fever in Nigeria: the high price of poor medical practice. Br Med J 311; 857-859.
- Fichet-Calvet E, Rogers DJ (2009) Risk map of Lassa fever in West Africa. PLOS Neglected Dis 3 (3) 1-13.
- Buchmeier MJ, Bowen MD, Peters CJ (2001) Arenaviridae: The viruses and their replication. In Fields Virology, 4th edition. Edited by: Knipe DM, Howley PM. Philadelphia: Lippincott-Raven; 1635- 1668.
- Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W (2001) The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc Natl Acad Sci U S A A98(22):12701-12705.
- Hass M, Westerkofsky M, M?ller S, Becker-Ziaja B, Busch C, G?nther S (2006) Mutational analysis of Lassa virus promoter. J Virol 12414-12419.
- Gallaher WR, DiSimone C, Buchmeier MJ (2001) The viral transmembrane superfamily: Possible divergence of arenavirus and filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol 1:1. 38:2670-2677.
- Carrion Jr. R, Brasky K, Mansfield K, Johnson C, Gonzales M, Ticer A, Lukasshevich I, Tardif F, Patterson J (2007) Lassa virus infection in experimentally infected marmosets: liver pathology and immunophenotypic alterations in target tissues. J Virol 6482- 6490.
- Lelke M, Brunotte L, Busch C, G?nther S (2010) An N-terminal region of Lassa virus L protein plays critical role in transcription but not replication of viral genome. J Virol 1934-1944.
- Rojek JM, Sanchez AB, Nguyen NT, de la Torre J, Kunz S (2008) Different mechanism of cell entry by human-pathogenic old world and new world arenaviruses. J Virol 7677-7687.
- Kern?is S, Koivogui L, Magassouba N, Koulemou K, Lewis R, Aplogan A, Grais RF, Guerin PJ, Fichet-Calvet E (2009) Prevalence and risk factors of Lassa virus seropositivity in inhabitants of the forest region of Guinea: a cross-sectional study. PLOS Neglected Trop Dis 3 (11) 1-9.
- Bowen MD, Rolling PE, Kisiazek TG, Hustand HL, Baush DG, Demby AH, Bajani MD, Peters CJ, Nichol ST (2000) Genetic diversity among lassa virus strain. J Virol 74 (15); 6992-7004.
- Takeda N, M Tanimura and K Tiyamura (1994) Molecular evolution of major capsid protein VP1 of enterovirus 70. J. Virol 844-862
- Okoror LE, Alaiya HT, Imarenezor KP, Aliga P, Erimafa JT (2007) Molecular evolution of lassa virus glycoprotein. Schriftenreihe Informatik 2007, 26: 21-29.
- Miyamura K, M Tanimura, N Takeda, R Kono, S Yamazaki (1986) Evolution of enterovirus 70 in nature: all isolates were recently derived from a common ancestor. Arch. Virol 89: 1-14.
- Banseh DG, Rollin PE, Demby AH, Coulibally M, Karu J, Conteh AS (2000) Diagnosis and clinical virology of lassa fever as evaluated by enzyme-linked immunosorbent assay. Indirect fluorescence antibody test and virus isolation. Clinical Microbiol 38. 2670-77.
- Gorman O T, W J Bean, Y Kawaoka, R G Webster (1990) Evolution of the nucleoprotein gene of influenza A virus. J. Virol 64:1487- 1497.
- Shu, L L, W J Bean, and RG Webster (1993) Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J. Virol 67:2723-2729.
- Tamura K, Dudley J, Nei M and Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution 10.1093/molbev/msm092
- Kamagae Y, S Sugita, A Endo, M Ishida, S Senya, K Osako, K (1994) VOL. 68.
- Yamashita M, M Krystal, W M Fitch, and P Palese (1998) Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virol 163:112-122.
- Gojobori, T, W.-H. Li, and D Graur (1982) Patterns of nucleotide substitution in pseudogenes and functional genes. J. Mol. Evol 18:360-369.
- Li, W.-H., C.-I. Wu, and C.-C. Luo (1985) A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol 2:150-174.
- Jahrling PB and Peters CJ (1986) Serology and Virulence diversity among Old World Arenaviruses and relevance to vaccine development. Med. Microbiol. And Immunol (175 2-3) 65-7.
- Geisbert TW, Jones S, Fritz EA, Shurtleff AC, Geisbert JB, Liesbscer AG et al. (2005) Development of a new vaccine for the prevention of Lassa fever. PLOS Med 2 (6) 0537-0545.
- Cosset F, Marianneau P, Verney G, Gallais F, Tordo N, Percheur E et al. (2009) Characterization of Lassa virus cell entry and neutralization with lassa virus pseudoparticles. J Virol 3228-3237.

