Introduction
Gemtuzumab Ozogamicin (“Gemtuzumab” or “Mylotarg”) binds to CD33 which is expressed on Acute Myeloid Leukaemia (“AML”) cells, but not on normal mature haematopoietic stem cells.The United States of America (“US”) Food and Drug Administration (“FDA”) approved the drug in 2000 via an accelerated licensing process for patients older than 60 years of age and after first relapse in AML. Mylotarg was withdrawn from the market in June 2010 at the request of the FDA when a clinical trial showed that the drug increased patient death and added no benefit over conventional therapies [1]. However, more recent research indicates that the drug may have a role in different patient populations with AML. Phase III clinical trials suggest that low and frequently repeated doses together with standard chemotherapy may be a viable option for the treatment of older adults aged 50 to 70 years of age [2]. Currently, Mylotarg is not commercially available to new patients outside of clinical trials and any future wider use will require resubmission of an investigational drug application to the FDA [1].
Risk governance & regulation
Governance, Risk and Compliance
Governance describes the principles and approaches employed to manage the control of organisations and systems. Its mechanisms aim to move away from a top-down approach towards those of more self-regulation. That said the framework for Governance is most often constructed from sets of rules, conventions based on best practice, and regu- latory and legal frameworks that encompass areas such as Safety, Finance, Information, and Environment. It is critical to the practice of organisations and systems that they operate within these codes (Compliance) and that Governance reporting is complete, accurate and timely in order to enable good decision-making. Risk management is the process by which organizations record, analyse, and respond to these concerns e.g., technology risks, commercial concerns, compliance with regulation, information security, product safety etc. Effective integrated Governance, Risk and Compliance monitoring is an essential component of Pharmaceutical business. They are the ‘glue’ holding together Quality, Performance, and statutory obligations [3].
Guidelines for Monoclonal Antibody (mAb) production
At every stage of drug development and dissemination regulators and pharmaceutical companies need to assess whether existing Good Laboratory Practice (“GLP”), Good Manufacturing Practice (“GMP”) and International Conference of Helsinki Good Clinical Practice (“ICHGCP”) guidance is adequate for safety compliance, and whether new guidance / legislation is required for new technologies, processes, or amendments to a process. Detailed regulatory guidelines for mAbs have been developed and revised since the early 1990s, constitute the framework for governance and riskassessment ‘tools’ for manufacture and distribution of all biological therapies, and are described in detail in World Health Organization, FDA, and European Union guidelines [4-9].
The ‘Critical Path’ - drug development to distribution
The ‘Critical Path’ to distribution of any new therapeutic agent includes the appropriate assessment and monitoring of:
i. Analytical Methods,
ii. Processes,
iii. Technologies,
iv. Manufacturing,
v. Clinical trials, and
vi. Distribution.
Table 1 breaks these components down in to discrete areas applicable to mAb technology, all of which require definition, analysis, recording and reporting, and through risk registration details of actions / mitigations in order to maintain compliance, safety, and quality.
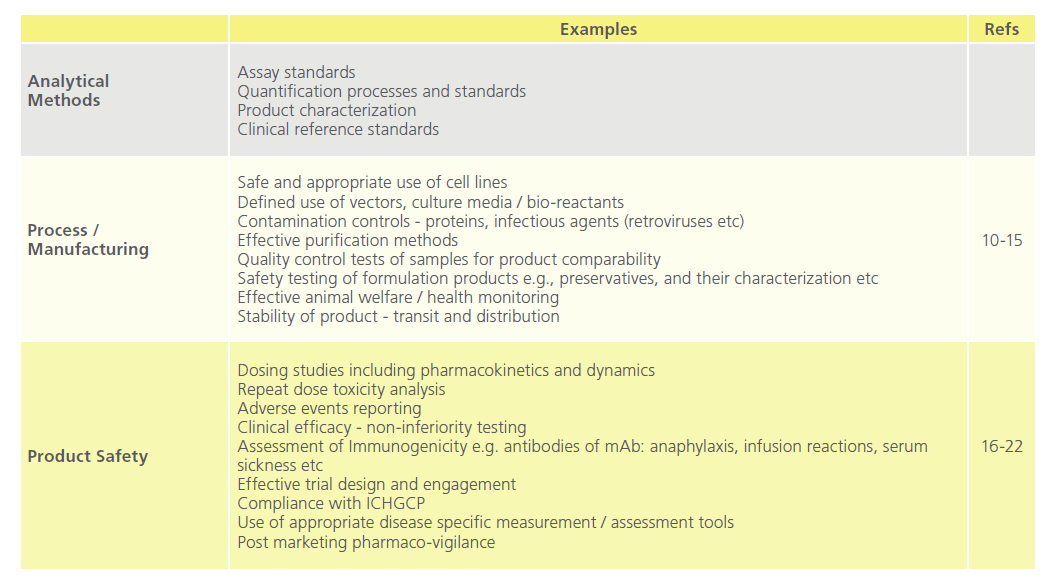
Table 1: Key Elements in the Critical Path to Drug Distribution.
The development of mAbs remains an evolving field in therapeutics. Structurally, they have evolved from fully murine mol- ecules to chimaeric, humanized and fully human molecules. Small changes in technology and processes can have considerable consequences. It is essential that non-clinical as well as clinical risk identification remain proactive [23].
Regulatory Background - EU perspectives
Approval of new drugs for human use are regulated by national laws [24] (Table 2). In the UK, once a product is regarded as medicinal by the Medicines and Healthcare products Regulatory Agency (“MHRA”) pursuant to the Medicinal Products Directive [25], the product is among others subject to the Medicines for Human Use Marketing Authorisations Regulations 1994 [26], and the Medicines Act 1968 [27-29].
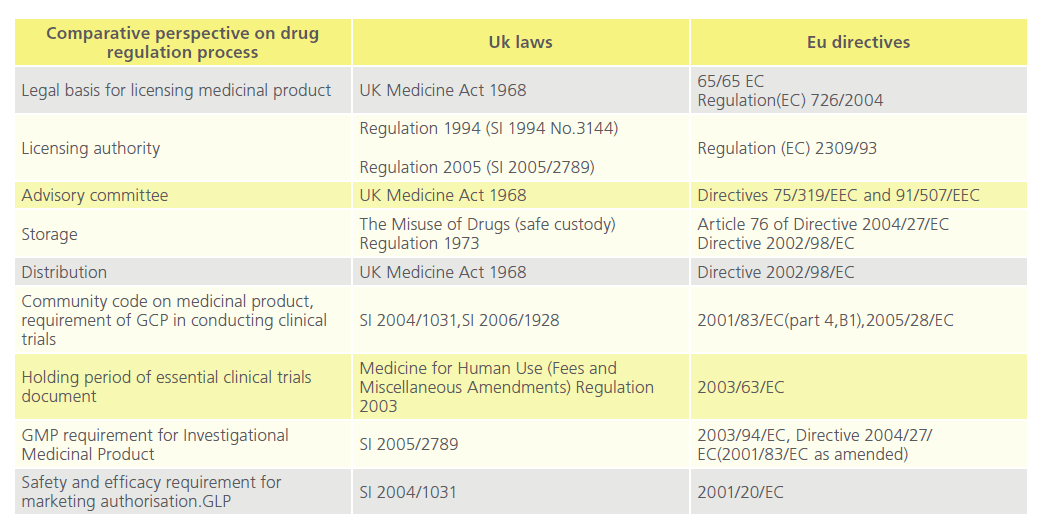
Table 2: Comparative perspectives on drug regulation process in the UK and the EU.
Marketing authorisation of a new drug comprises of various stages: application to conduct clinical trials, conducting clinical trials, application to marketing authorisation of drug and post-marketing studies. Every country has its own regulatory authority, which is responsible for enforcing the rules and regulations and issuing marketing guidelines.Table 3 compares the processess involved before a marketing authorisation is given in the US and EU [30].
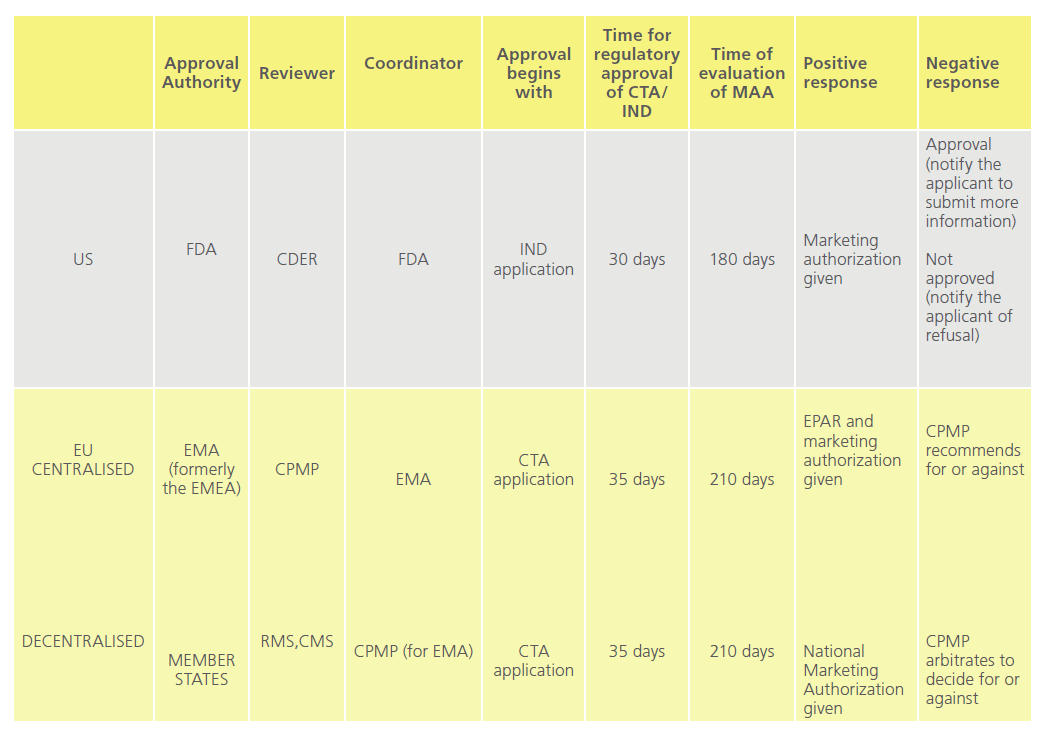
Table 3: Comparison of new drug approval in the US and EU.
Orphan drug regulation
The US Orphan Drug Act [31] was designed to facilitate the development and commercialisation of drugs to treat rare diseases. The enforcement of this encouraged other countries and unions such as Australia, Japan, Singapore and the EU to take respective regulatory measures in this field [32-34]. Table 4 compares orphan drug policy in the EU with that of the US.
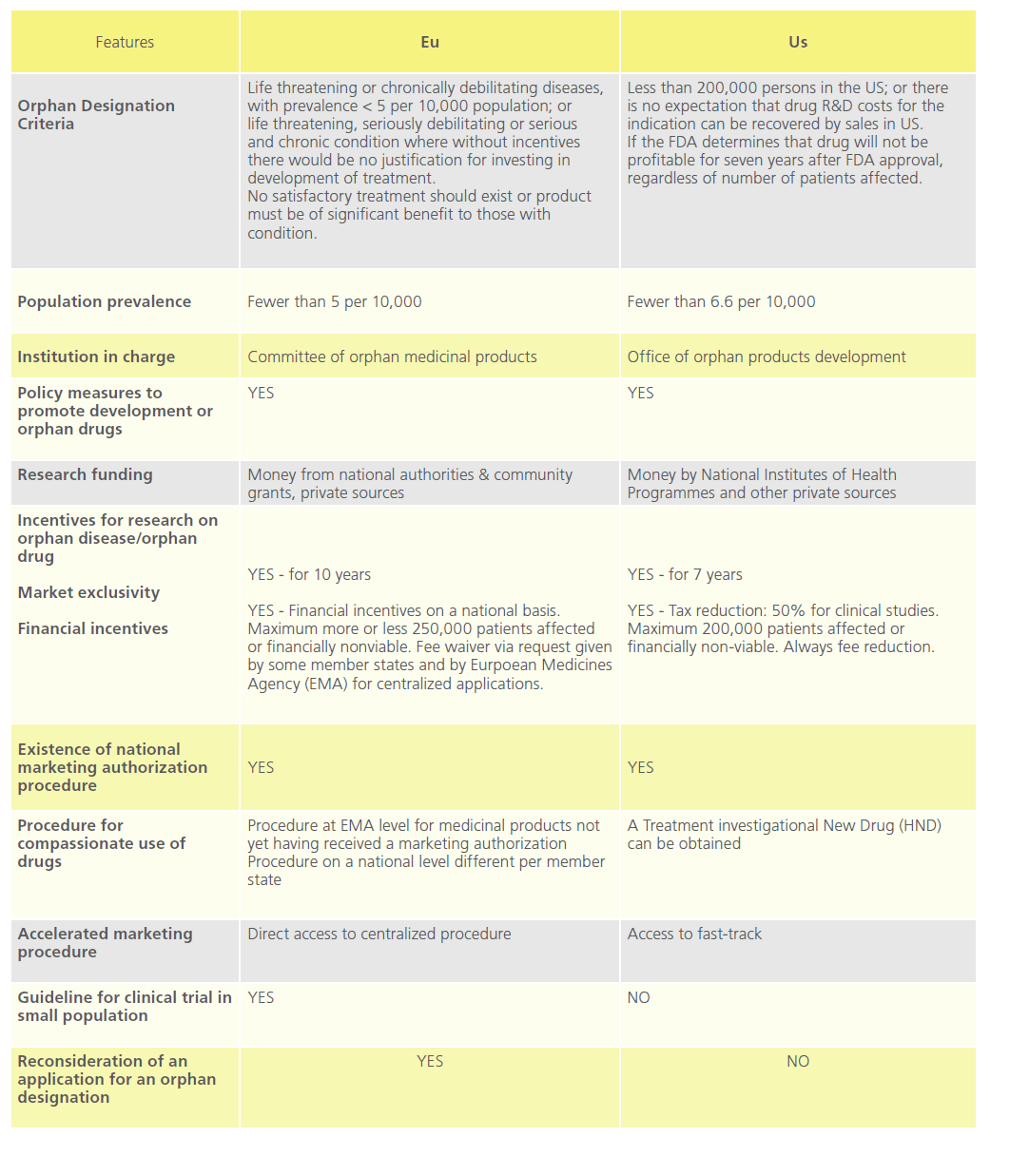
Table 4: Comparison of EU and US in the field of Orphan drugs.
The MHRA has not been involved with orphan drug marketing authorisation to date. Orphan drugs in the UK are authorised through the centralised procedure at European Medicines Agency (“EMA”, formerly the “EMEA”) through the Committee for Orphan Medicinal Products [35,36]. Designated orphan medicinal products are investigational products which are considered for designation on the basis of potential activity [37]. An orphan designation is not a marketing authorisation, as such demonstration of the quality, safety and efficacy will be necessary before a product can be granted a marketing authorisation [38].
Box 1. Mylotarg’s Marketing Approval
Marketing approval of Mylotarg was granted on May 17, 2000 by the FDA under the Accelerated Approval regulations. Orphan designation (EU/3/00/005) was granted on 18 October 2000 to Wyeth Europa Limited United Kingdom for the treatment of AML. The application contained a critical report addressing the possible similarity with authorised orphan medicinal product Trisenox. However, in 2007, its application for authorisation in Europe was rejected by the EMA due to scarcity of data and toxicities concern [39].
CTA: Clinical Trial Application, CDER: Centre for Drug Evaluation and Research, IND: Investigational New Drug, EPSAR: European public Assessment Report, RMS: Reference Member State, CMS: Concerned Member State, MAA: Marketing Authorization Application, FDA: Food and Drug Administration, EM(E)A: European Medicines (Evaluation) Agency, CPMP: Committee for Proprietary Medicinal Products.
Clinical trials
The EU Clinical Trials Directive [40] was introduced to establish standardisation of research activity in clinical trials throughout the European Community [41,42]. It was transposed into the UK law as the Medicines for Human Use Regulations, and included the following additional controls:
i. Establishment of ethics committees on a legal basis
ii. Each clinical trial must have an identified sponsor taking responsibility for its initiation, conduct and management
iii. Phase 1 pharmacology studies in healthy volunteers require authorisation by the MHRA, and
iv. Investigational medicinal products (IMPs) must be manufactured to GMP standards and the manufacturer must have a manufacturing licence [43,44].
These also mandate the MHRA to inspect, enforce and set standards to ensure quality in the manufacturing and supply of medicines on the UK market while the Good Clinical Practice Directive [45] supplements the Clinical Trials Directive, strengthening the legal basis for requiring Member States to comply with the principles and guidelines of good clinical practice. The community code relating to medicinal products for human use stipulates that clinical trials data used for marketing authorisation applications in the EU shall be conducted in accordance with Good Clinical Practice (“GCP”)[45].
Box 2. Mylotarg’s Trials
The pharmacology of Mylotarg was investigated both in vitro and in vivo. Pharmacodynamic and pharmacokinetic studies were not conducted according to GLP standards. The toxicity studies in rats and cynomolgus monkeys and the reproductive and developmental toxicity studies in rats were conducted according to GLP standards.The human clinical trials were carried out in accordance with the community code relating to medicinal products for human use. The clinical programme comprised three phase I/II dose-finding studies (0903A1-101- US, 0903A1-102-US, and 0903A1-103-JP) and three phase II, open-label, single-arm, 3-part, multidose, multicentre clinical trials (0903B1-201-US/CA, 0903B1-202-EU, and 0903B1- 203-US/EU). A total of 377 patients were screened and 277 patients were evaluated for efficacy after the pooling of studies. The study was carried out in 3 parts: in part one 277 enrolled and received dose one, 210 received dose two, and 7 received dose three. Of these 44 people died. In part two 223 entered 6-month follow-up and 128 died. In part 3,105 entered 18-month follow-up, and 80 died while 25 remain in part three [46]. (Table 5 and Table 6)
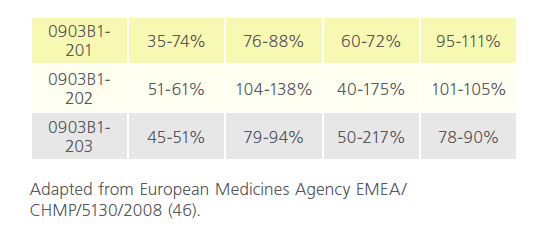
Table 5: Summary of between-subject case variation in studies 0903B1-201-US/CA, 0903B1-202-EU, and 0903B1-203-US/EU (data for hP67.6
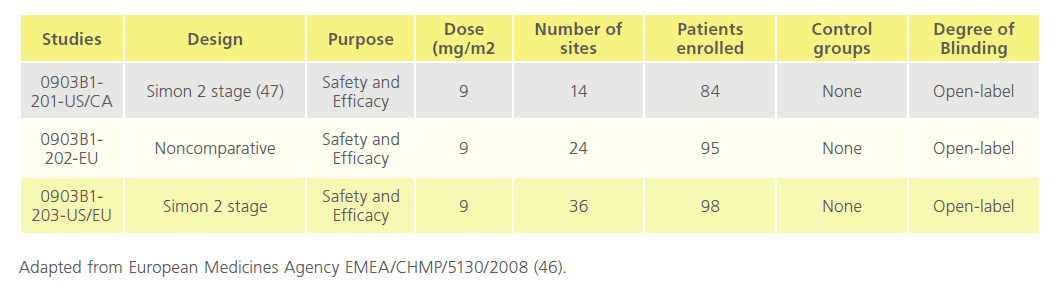
Table 6: Overview of studies 0903B1-201-US/CA, 0903B1-202-EU, 0903B1-203-US/E.

Table 7: Strategic aspects in antibodypa
Good manufacturing practise
The EU Manufacturing Directives [45,48] set the principles and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human use.
Box 3. Manufacturing Mylotarg
Mylotarg was manufactured by an approved manufacturer in compliance with GMP. Subsequent to an EMA request during evaluation, an in-process control test on residual moisture on the lyophilised powder was introduced and the limits for the in-process controls performed during lyophilisation (i.e. temperature, time and pressure) have been stated. Given the drugs limited stability it is shipped to the lyophilisation site immediately after manufacture prior to the completion of all testing and formal release. Release of the resulting drug product batches is contingent upon acceptable results for drug substance release testing.
Commercialisation and intellectual property
Advertising and promotion
The regulation of Direct-to-Consumer (DTC) advertising of prescription drugs substantially differs among jurisdictions. In the US such advertising is generally allowed under FDA supervision [49], whereas in the EU it is forbidden. EU Directives on advertising of medicinal products for human use set criteria for advertising of medical products [50]. Regulation is complemented by industry codes of conduct such as those of the European Federation of Pharmaceutical Industries and Associations (EFPIA) [51].
DTC Advertising within the EU
Within the EU among others the following is prohibited:
i. Advertising to the general public of medicinal products which are only available on medical prescription;
ii. Mentioning, when advertising to the general public, therapeutic indications where self-medication is not suitable, and
iii. The distribution of free samples, gifts or bonuses to the general publi [50].
Also, authorised advertising to the general public must:
i. Be set out in such a fashion that it is clear that the message is an advertisement, and that the product is clearly identified as a medicinal product;
ii. Include all the necessary information for correct administration of the medicinal product;
iii. Include an express invitation to read the instruction leaflet carefully, and
iv. Not include elements incompatible with the rational administration of the medicinal product [50]
Tension exists between commercial advertising that focuses materials on helping consumers acquire more information about diseases and prescription drugs, facilitating consultation with doctors (a pharmaceutical company perspective), versus information that should clarify who is at the high risk, and the risk-benefit balance (a regulator perspective)[52].
Advertising to professionals within the EU
Any advertising to professionals must include essential information compatible with the summary of the product’s characteristics and the classification of the medicinal product for supply purposes. Inducements to prescribe or supply medicinal products are forbidden, and the supply of free samples to persons qualified to prescribe or supply medicinal products is subject to strict controls [50,51]. Furthermore, pharmaceutical companies are required to establish within the company a scientific service in charge of information relating to medicinal products [50].
Post-marketing surveillance
Beyond phase III clinical trials further attempts are made to assess safety and efficacy in broader settings of routine clinical practice. Data capture goes beyond adhoc reporting of complications, using more structured post-marketing surveillance, essentially in uncontrolled cohort trials most often funded by the pharmaceutical company but managed by regional or national clinical groups such as specialty organisations or clinical research bodies. Comparison of this data with controlled clinical trials may help obtaining regulatory approval [53,54], identify inferiority / superiority over other treatment options, and inform further study opportunities within care pathways appropriate to patient subgroups not necessarily identified (even originally excluded) from the clinical trials.
Recent guidance from regulatory agencies such as the FDA and EMA emphasises the use of quantitative clinical evidence of risk, but does not address the value of therapeutic benefits to patients, physicians or other stakeholders. The willingness of stakeholders to trade off risks for benefits has been studied using approaches such as incremental net health benefits (“INHB”) and maximum acceptable risk (“MAR”) analyses. These assessments can be conducted at any stage in development and dissemination of new treatment
In the INHB approach both benefits and adverse events associated with a new treatment are quantified by using postmarketing surveillance data. Weighted scores are assigned to every outcome and analysed against reference measures from appropriate comparators. A positive INHB indicates the net benefits of treatment are superior to a comparator. The main purpose of MAR is to estimate the maximum risk patients are willing to accept in order to achieve different levels of benefit from a new therapy [56]. A new treatment is deemed acceptable or not by comparing MAR against the actual or expected risk. Both MAR and INHB are promising approaches to comparing risks and benefits of new and current therapeutic methods.
Intellectual Property (IP)
Protection of IP is a key component of pharmaceutical company business strategies. Patents, ‘know-how’ and regulatory exclusivity are among the most relevant forms of IP arising from pharmaceutical research and development (“R&D”). During R&D activities, a great deal of confidential information may accumulate, both for registration purposes and through the general handling of the medicine. Thus, it is important that the originator keeps such information secret as once it is published in detail competitors can use it to help obtain their own health registrations. Employee invention policies regulating among others notification obligations, disclosure issues, and the company’s incentive mechanisms are necessary tools for R&D governance. Comprehensive non-disclosure agreements are also highly important for any pharmaceutical company engaging with internal or external resources in its R&D activities.
Patenting monoclonal antibodies
To meet patentability criteria [59] a mAb must:
i. Be novel (i.e., not disclosed in single prior art reference, EPC Article 54)
ii. Be nonobvious (i.e., cannot be obvious modification of what is already publicly known in field EPC Article 56)
iii. Describe antibody with sufficient detail (enough to show that inventors had possession of full scope of what is claimed as invention, EPC Article 83)
iv. Enable how to make and use antibody (EPC Article 100(b))
v. Be industrially applicable (EPC Article 57), and
vi. Teach best mode known to inventors of making/using invention (the US only).
Disclosure, novelty and nonobviousness assessment
When prosecuting mAb patent applications before the European Patent Office (“EPO”) the scope of protection and acceptable breadth of the patent claims depends on the detail of the disclosure of the claimed protein or peptide in the description of the application [58]. When it comes to claims directed to mAbs, the minimum structural information necessary would be the heavy and light chain variable region sequences of the antibody, or at least the sequences of the complementarity determining regions of the heavy and light chain [60]. One variable region/chain or less than 6 Complementary Determining Regions (“CDR”) is not enough, as a normal heavy/light chain alone does not bind sufficiently and does not have the technical effect shown for the entire antibody (i.e., lack of inventive step). Single CDRs i.e. peptides, are not usually novel. However, in exceptional cases (single domain antibodies or binding peptides) an inventive step can be acknowledged if experimental data is provided [60]. The approach applied by United States Patent and Trademark Office (“USPTO”) is nearly identical. If sufficient sequence determinants are included in the claim, the EPO will often grant protection of the antibody as such. However, patent protection is thereby limited to antibodies containing exactly these structural features [60].
To acquire a broader scope of protection, the applicant may try to claim antibodies or proteins with a minimum identity to specific sequences of the antibody or protein. In such case, the application may disclose several sequences with modifications in one or more amino acids, all providing the same desired technical effect. Generalisations in claims directed to antibodies are acceptable only if the solution of the problem underlying the invention was made credible over the full range of claimed antibodies or proteins [60].
Many pioneering antibody technologies are now mainstream (humanisation, phage display, transgenic mice etc). Furthermore, patent protection for the basic protein is generally pursued very early in the R&D process; this and the extensive regulatory review timeframe means that a significant period of the patent term has been lost by the time of product launch.
Box 4. Mylotarg’s registered IP
Searches in public patent databases reveal that a US patent has been granted for “Combination therapy for the treatment of acute leukemia and myelodysplastic syndrome” to Wyeth LLC. The patent was granted on 1 June 2010 (filed on 11 June 2007). The abstract of the patent describes the invention as “methods of treatment and pharmaceutical combinations are provided for the treatment of acute leukemia, such as acute myelogenous leukemia, and myelodysplastic syndrome”. The methods of treatment and pharmaceutical combinations employ an anti-CD33 cytotoxic conjugate in combination with at least one compound selected from the group consisting of an anthracycline and a pyrimidine or purine nucleoside analog. Preferred methods of treatment and pharmaceutical combinations employ gemtuzumab ozogamicin, daunorubicin, and cytarabine.” [61,62]. Gentuzumab has been marketed under the Mylotarg® brand, a registered trademark owned by Wyeth LLC [63].
Summary
This chapter has outlined the key regulatory and compliance aspects for drug development and dissemination. This complex set of rules ranges from pre-clinical laboratory and manufacturing standards, through intellectual property and marketing regulations, to clinical safety. Although Mylotarg has passed through the majority of these regulations in development as a mAb, it now requires clinical trial data ensuring safety and efficacy prior to resubmission of an investigational drug application and further consideration of its marketing strategy and post-marketing clinical surveillance.
Acknowledgement
We wish to thank Prof.Roy Douglas(Programme Director and Member of the Development Team) for his supervisory role in this paper.
5833
References
- 1 FDA’s News Release: Mylotarg (gemtuzumab ozogamicin): Market Withdrawal, dated 21June 2010, available at: https:// www.fda.gov/Safety/MedWatch/SafetyInformation/ SafetyAlertsforHumanMedicalProducts/ucm216458.htm
- Azvolinsky A . Conference Report: ASH: Gemtuzumab- A Potential New Use for an Old Drug in AML, available at: https:// www.cancernetwork.com/conferencereports/ash2011/content/ article/10165/2005126
- Tarantino A. Governance, Risk, and Compliance Hanbook. 2008. John Wiley & Sons. Hoboken, New Jersey.
- van de Donk HJM et al. (1991 Geneva Committee). Guidelines for assuring the quality of monoclonal antibodies for humans. WHO. 1992; 822:48-68
- Zoon KC. Manufacturing and testing of monoclonal antibodies. US Dept. of Health and Human Science. Food and Drugs Agency. Centre for Biologics Evaluation and Research. 1997, February.
- Guidance for Industry. Interpreting sameness of monoclonal antibody products under the orphan drug regulation. US Dept of Health and Human Science. Food and Drugs Agency. Centre for Biologics Evaluation and Research. Centre for Drugs Evaluation and Research. 1999, July.
- Guideline on similar biological medicinal products containing monoclonal antibodies. EU Medicines Agency. 2010, November.
- Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: quality issues. EU Medicines Agency reference 49348/05.
- Guideline on production and quality control of monoclonal antibodies and related substances. EU Medicines Agency reference 157653/07
- Snodin DJ, Ryle PR. Understanding and applying regulatory guidance on the nonclinical development of biotechnology-derived pharmaceuticals. BioDrugs. 2006;20(1):25-52.
- Celis P, Silvester G. European Regulatory guidance on virus safety of recombinant proteins, monoclonal antibodies and plasma derived medicinalproducts. Dev Biol (Basel). 2004;118:3-10.
- Levine HL, Jagohies G. The development of therapeutic monoclonal antibody products. A comparative guide to CMC activities from clone to clinic. BioProcess Technology Consultants and GE Health. 2010. 330 pages.
- Jones SD, Seymour P, Levine HL. Chemical Manufacturing Control (CMC) activities for development of drugs. Critical steps to reach Investigative New Drug (IND) application status with a therapeutic antibody. Contract Pharma. 2010;4:60-3.
- Lynch CM, Hart BW, Grewal IS. Practical considerations for nonclinical safety evaluation of therapeutic monoclonal antibodies. MAbs. 2009; 1(1):2-11.
- Lynch CM, Grewal IS. Preclinical safety evaluation of monoclonal antibodies. Handb Exp Pharmacol. 2008;181:19-44.
- Klingbeil C, Hsu DH. Pharmacology and safety assessment of humanized monoclonal antibodies for therapeutic use. Toxicol Pathol. 1999;27(1):1-3.
- Tabrizi MA, Roskos LK. Preclinical and clinical safety of monoclonal antibodies. Drug Discov Today. 2007;12(13-14):540-7.
- Chapman K, Pullen N, Graham M, Ragan I. Preclinical safety testing of monoclonal antibodies: the significance of species relevance. Nat Rev Drug Discov. 2007 Feb;6(2):120-6.
- Muller PY, Brennan FR. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin Pharmacol Ther. 2009 Mar;85(3):247-58.
- Dixit R, Coats S. Preclinical efficacy and safety models for mAbs: the challenge of developing effective model systems. IDrugs. 2009;12(2):103-8.
- Brennan FR, Morton LD, Spindeldreher S, Kiessling A, Allenspach R, Hey A, Muller PY, Frings W, Sims J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs. 2010;2(3):233-55.
- Liedert B, Bassus S, Schneider CK, Kalinke U, Löwer J. Safety of phase I clinical trials with monoclonal antibodies in Germany--the regulatoryrequirements viewed in the aftermath of the TGN1412 disaster. Int J Clin Pharmacol Ther. 2007;45(1):1-9.
- Schneider CK. Monoclonal antibodies--regulatory challenges. Curr Pharm Biotechnol. 2008;9(6):431-8.
- Dukes MNG. OECD International futures project on “the bioeconomy to 2030: Designing a Policy Agenda” biotechnology regulation in the health sector. 2008. Available at https://www. oecd.org/dataoecd/11/14/40926707.pdf*https://www.oecd.org/ dataoecd/11/14/40926707.pdf
- Directive 2001/83/EC of the European Parliament and Council of 6 November 2001 on the Community Code Relating to Medicinal Product for Human Use.Available at: https://europa.eu/ legislation_summaries/internal_market/single_market_for_goods/ pharmaceutical_and_cosmetic_products/l21230_en.htm
- The UK Medicines for Human Use Marketing Authorisations Regulations 1994. Available at: https://www.legislation.gov.uk/ uksi/1994/3144/contents/made\|https://www.legislation.gov.uk/ uksi/1994/3144/contents/made
- The UK Medicines Act, 1968. Available at: https://www.legislation.gov. uk/ukpga/1968/67\|https://www.legislation.gov.uk/ukpga/1968/67
- Cohen J, Cairns C, Paquette C, Faden L. Comparing patient access to pharmaceuticals in the UK and US Appl Health Econ Health Policy. 2006;5(3):177-87.
- McGoey L, Jackson E. Law, ethics and medicine. Seroxat and the suppression of clinical trial data: regulatory failure and the uses of legal ambiguity. J Med Ethics. 2009;35:107-12.
- Dulichand R, Harish D. New drug approval process: regulatory view. Pharmaceutical Reviews. 2010; 8(4). Available at: https://www. pharmainfo.net\|https://www.pharmainfo.net
- The US Orphan Drug Act of 1983. Available at: https://www.fda.gov/regulatoryinformation/ legislation/federalfooddrugandcosmeticactfdcact/ significantamendmentstothefdcact/orphandrugact/ default.htm\|https://www.fda.gov/regulatoryinformation/ legislation/federalfooddrugandcosmeticactfdcact/ significantamendmentstothefdcact/orphandrugact/default.htm
- Shah RR. Regulatory framework for the treatment of orphan diseases. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis. 2006.
- Sharma A, Jacob A, Tandon M, Kumar D. Orphan drug: development trends and strategies. J Pharm Bioallied Sci. 2010;2(4):290-9.
- Tambuyzer E. Rare diseases, orphan drugs and their regulation: questions and misconceptions. 2010;9(12):921-9
- Regulation (EC) No 141/2000 of the European Parliament and Council of 16 December 1999 on orphan medicinal products. Available at: https://europa.eu/legislation_summaries/internal_market/single_ market_for_goods/pharmaceutical_and_cosmetic_products/l21167_ en.htm
- Denis A, Simoens S, Fostier C, Mergaert L, Cleemput I. Policies for orphan diseases and orphan drugs. health technology assessment (HTA). Brussels: Belgian health care nnowledge centre(KCE); 2009. KCE reports 112C (D/2009/10.273/32). Available at: https://ec.europa. eu/health/ph_threats/non_com/docs/policies_orphan_en.pdf
- Roberta J, Vittorio B, Silvio G. Orphan drug development is not taking off. Br J Clin Pharmacol. 2009; 67(5):494-502.
- National Research Council. Rare diseases and orphan products: accelerating research and development. Washington, DC: The National Academies Press, 2011;73-110.
- Bross PF, Beitz J, Chen G et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490-6.
- Directive 2001/20/EC of the European Parliament and Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Available at: https://ec.europa. eu/health/human-use/clinical-trials/index_en.htm.
- Duncan N. Acute myeloid leukemias: Current and future treatments. Clinical Pharmacist. 2010;2:123-7.
- The UK Medicines for Human Use Regulations (Clinical Trials) Regulations 2004 (SI 2004/ 1031). Available at: https://www.legislation. gov.uk/uksi/2004/1031/contents/made
- Hartmann M, Hartmann-Vareilles F. The clinical trials directive: how is it affecting Europe’s noncommercial research? PLoS Clin Trials. 2006;1(2):e13.
- Flavell DJ , Flavell SU, Sullivan R. European clinical trials directive: responses made to MHRA consultation letter MLX 287. The Lancet. 2003;362(9393):1415
- Commission Directive 2005/28/EC of 8 April 2005 laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products. Available at: https://ec.europa.eu/health/documents/ eudralex/vol-1/index_en.htm
- European Medicines Agency: Pre-authorisation Evaluation of Medicines for Human Use: Refusal asessment report for mylotarg.Doc. ref.: EMEA/CHMP/5130/2008, Procedure No.EMEA/H/C/000705.
- Simon R. Optimal two stage designs for phase II clinical trials. Controlled Clinical Trials. 1989;10:1-10.
- Directives 91/356/EEC, as amended by Directive 2003/94/EC, and 91/412/EEC respectively. Available at: https://ec.europa.eu/health/ documents/eudralex/vol-4/index_en.htm
- Rados C. Truth in advertising: Rx drug ads come of age. FDA Consum. 2004;38:20-7.
- Council Directive 92/28/EEC of 31 March 1992 regarding advertising of medicinal products for human use, commentary and directive. Available at: https://europa.eu/legislation_summaries/other/l21143_ en.htm
- EFPIA HPC Code on promition of prescription only mdedicines to, and interactions with health care professionals. Available at: https://www. efpia.eu/Content/Default.asp?PageID=559&DocID=11731
- Frosch DL, Krueger PM, Hornik RC et al. Creating demand for prescription drugs: a content analysis of television direct-to-consumer advertising. Ann Fam Med. 2007;5:6-13.
- Montaner JS, O’Shaughnessy MV, Schechter MT. Industry-sponsored clinical research: a double-edged sword. Lancet. 2001;358:1893-5.
- Gordon J, Watson M, Avenell A. Lightening the load? A systematic review of community pharmacy-based weight management interventions. Obes Rev. 2011;12:897-911
- Sutton AJ, Cooper NJ, Abrams KR, et al. A Bayesian approach to evaluating net clinical benefit allowed for parameter uncertainty. J Clin Epidemiol. 2005;58:26-40.
- Ryan M, Scott DA, Reeves C, et al. Eliciting public preferences for healthcare: a systematic review of techniques. Health Technol Assess. 2001;5:1-186.
- Case Merck KGaA v. Integra Lifesciences I, Ltd. Available at: https:// www.law.cornell.edu/supct/html/03-1237.ZO.htmlhttps://www.law. cornell.edu/supct/html/03-1237.ZO.html.
- In the EU corresponding exemptions are allowed under the terms of EC Directives 2001/82/EC (as amended by Directive 2004/28/EC) and 2001/83/EC (as amended by Directives 2002/98/EC, 2003/63/EC, 2004/24/EC and 2004/27/EC). Available at: https://www.mhra.gov.uk/ Howweregulate/Medicines/ReviewofEUmedicineslegislation(2001Revi ew)/index.htm
- The European Patent Convetion. Available at: https://www.epo.org/ law-practice/legal-texts/epc.html
- Minderop R, Burrichter A, Diepholz M, Kirchhofern N. Patenting Biotechnology inventions via the EPO. Patents in Europe 2011/2012, 15-19. Available at: www.iam-magazine.com.
- US Patent no: 7,727,968: “Combination therapy for the treatment of acute leukemia and myelodysplastic syndrome”. Available at USPTO patent database: https://patft.uspto.gov/netacgi/nphParser?Sect2=P TO1&Sect2=HITOFF&p=1&u=/netahtml/PTO/search-bool.html&r=1& f=G&l=50&d=PALL&RefSrch=yes&Query=PN/7727968https://patft. uspto.gov/netacgi/nphParser?Sect2=PTO1&Sect2=HITOFF&p=1&u=/ netahtml/PTO/search-bool.html&r=1&f=G&l=50&d=PALL&RefSrch=ye s&Query=PN/7727968 .
- EPO’s patent database, gemtuzumab patent applications. Available at: https://worldwide.espacenet.com/publicationDetails/inpadocPatentFa mily?CC=US&NR=2004152632A1&KC=A1&FT=D&ND=5&date=2004 0805&DB=worldwide.espacenet.com&locale=en_EPhttps://worldwide. espacenet.com/publicationDetails/inpadocPatentFamily?CC=US&NR=2 004152632A1&KC=A1&FT=D&ND=5&date=20040805&DB=worldwi de.espacenet.com&locale=en_EP.
- Mylotarg trademark no: 85440982. Available at: https://trademarks. justia.com/854/40/mylotarg-85440982.htmlhttps://trademarks.justia. com/854/40/mylotarg-85440982.html tentingtion.

