Keywords
|
| Myelodysplastic syndrome; Anemia; Anisocytosis; Peripheral blood smear |
Introduction
|
| Myelodysplastic syndromes (MDS) encompass a diverse set of disorders that affect hematopoiesis in the bone marrow leading to cytopenia in red cells, myeloid cells, and platelets in the peripheral blood. MDS is usually diagnosed upon exclusion of other conditions and documentation of clonal cytogenetic or chromosomal abnormalities and/or increased myeloblasts in the bone marrow [1]. Cytopenia is often first seen in a complete blood count test and follow-up tests to determine the primary cause include a peripheral blood smear, flow cytometry, MDS FISH, and a bone marrow biopsy. MDS can present heterogeneously within the population and has been linked to multiple different genetic abnormalities, including silencing genes involved with tumor-suppression and DNA repair, all of which lead to inefficient hematopoiesis and abnormal myeloid lineage differentiation [2]. It has been shown that bone marrow from MDS patients has a higher fraction of hematopoietic stem cells with apoptotic characteristics than normal stem cells, suggesting hematopoiesis is unsuccessful due to a higher incidence of apoptosis [3]. Taken together, MDS patients exhibit decreased cell differentiation during hematopoiesis, increased progenitor cell apoptosis, and aberrant cell growth of a mutated myeloid lineage [4]. |
| MDS affects the normal process by which blood cells are produced in the bone marrow microenvironment following differentiation of hematopoietic stem cells into the common myeloid progenitor and specific erythroid, myeloid, and megakaryocytic lineages [5-8]. Clonal genetic aberrations in MDS cells lead to ineffective hematopoiesis with decreased numbers of differentiated cells in the marrow resulting in anemia, neutropenia, and thrombocytopenia in the blood. The diagnosis of MDS starts with a complete blood count and examination of the peripheral blood smear. The case presented below illustrates the significance of morphological abnormalities in the blood smear that are relevant to the diagnosis of MDS with a discussion of the pathogenesis of anemia and anisocytosis in MDS. |
Case Report
|
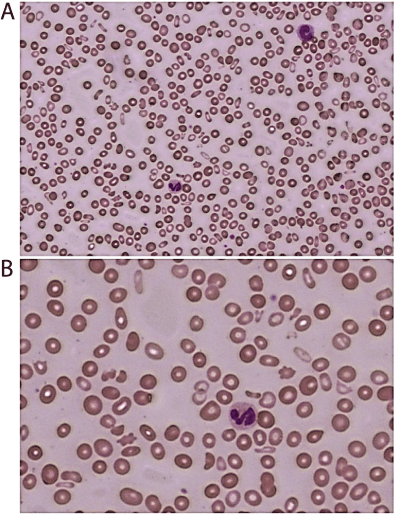
| A 65-year-old man was referred for evaluation of suspected myelodysplastic syndrome (MDS). He had been treated for severe anemia with blood transfusions prior to the office visit. |
| The complete blood count showed 5,400 leukocytes/μL, hemoglobin of 77 gm/l and a platelet count of 63,000/μL. The mean corpuscular hemoglobin concentration (MCHC) was low (32.1 g/dL) with a high red cell distribution width coefficient of variation (RDWCV) of 24.5% and RWD-SD at 76.4 fL. The peripheral blood smear confirmed the presence of anisocytosis (Figure 1), with the appearance of red cell fragments, hypochromic red cells, low platelets and early myeloid forms. A bone marrow biopsy and aspirate showed mild dyserythropoiesis, left shifted myeloid maturation and nuclear to cytoplasmic asynchrony, with atypical megakaryocytes, without excess blasts, consistent with early stage MDS. Cytogenetics and a MDS FISH panel showed a normal male karyotype, without chromosomal translocations commonly seen in MDS. Flow cytometry demonstrated that 8% of cells had an atypical monocytoid phenotype and expressed CD2 (dim), CD4 (dim), CD11b, CD11c, CD15, CD13, CD14, CD33, CD36, CD38, CD56, and CD45. Together, these results are consistent with the diagnosis of refractory cytopenia with multilineage dysplasia (RCMD), according to the 2008 World Health Organization (WHO) classification system [9]. Additional laboratory data included a normal LDH and a slightly elevated bilirubin at 1.3 mg/dL. The patient is currently receiving hypomethylating drug therapy and undergoing HLA typing to determine feasibility of allogeneic bone marrow transplantation. |
Discussion
|
| This patient presented with marked anemia and thrombocytopenia, indicating defects in erythropoiesis and thrombopoiesis. Normal erythropoiesis involves massive expansion of nucleated erythroid precursors in the marrow in the setting of a regulatory network of growth factors, cytokines, and cellular interactions. Erythropoietin produced by the kidneys in response to relative anemia and hypoxemia regulates the level of erythroid development in the marrow to maintain homeostatic levels of mature erythrocytes in the blood [8,10,11]. As hematopoietic stem cells differentiate down the erythroid lineage they generate common myeloid progenitors (CMP), megakaryocytic-erythroid progenitors (MEP), and BFU-E [12]. Progenitor cells differentiated beyond BFU-E are found in a colony-forming unit (CFU) as part of a larger erythroblastic island that enables late stage erythropoiesis [12]. In the bone marrow microenvironment, numerous erythroid cells, all between the BFU-E and enucleating erythrocyte stages, surround a macrophage. This macrophage increases the proliferation rate, phagocytoses the extruded nuclei, and recycles the resulting nucleic acids [12]. As erythrocytes are maturing, they usually go through approximately 3-5 cell cycles and accumulate iron to be used for hemoglobin synthesis [12]. In parallel, production of membrane proteins is also increased to enable terminal differentiation [12]. During these final differentiation stages, developing erythroblasts become smaller, condense their genetic material, extrude the nucleus, and degrade intracellular organelles to result in a deformable cell with a high surface area to volume ratio, enabling optimized oxygen delivery in the microvasculature [12]. Enucleation of erythroblasts generates reticulocytes that leave the bone marrow microenvironment and mature into erythrocytes in the blood. When erythrocytes age, they are cleared by the reticulo-endothelial system and their cytoplasmic constituents are recycled to the marrow for the biosynthesis of new erythroblasts. |
| The anisocytosis, or red blood cells that exhibit a broad size distribution including larger and smaller sizes than normal, seen in this patient is of interest as it may suggest the underlying DNA mutations and the specific subclass of MDS. In normal erythropoiesis, red blood cell size is regulated by the availability of the biochemical constituents needed for maturation of the nucleus and the cytoplasm during red cell development including heme, B12, folate, and protein. Reduction in cell size from erythroblasts to reticulocytes is normally achieved by reducing the duration of the G1 phase of the cell cycle while maintaining the same protein production rate. There are a multitude of mutations in MDS that can result in ineffective maturation of erythrocytes, altering the process of red blood cell development and enucleation [13]. Macrocytosis, or red blood cells larger than normal, is the result of a flaw in nuclear maturation, a relative lack of B12 or folate needed for DNA synthesis, or increased erythropoietin stimulation where atypically large cells are released to the bloodstream [14]. When cell cycle progression is delayed during erythroblast division, protein production continues over a longer cell cycle, creating a macrocyte with more protein and cytoplasm relative to the nucleus [15]. In MDS, mutations causing abnormal cytoplasmic and nuclear maturation in the final stages of erythrocyte differentiation often result in macrocytosis and anisocytosis as seen in the present case [16]. Microcytosis, or red blood cells smaller than normal in the blood smear, can result from decreased hemoglobin synthesis in an iron deficient environment, or sub-normal protein production leading to lower cytoplasmic volume relative to the nucleus and a smaller overall size [14,17]. Another possibility, suggested by the microcytosis and low mean corpuscular hemoglobin concentration in this case, is that this patient has developed α-thalassemia alongside MDS (ATMDS) [18]. It has been reported that ATMDS may be underreported as flow cytometry is not designed to monitor Hemoglobin H, but other methods have found Hb H in up to 8% of patients with MDS [18]. |
| Although cytogenetic abnormalities have been reported in 20-70% of patients upon diagnosis [19], the chromosomal translocations commonly seen in MDS were not observed with this patient, who had a normal male karyotype. In the absence of clonogenic chromosomal aberrations, the diagnosis of low-risk MDS can be somewhat subjective, with discrepancies seen in calculating the risk score between centers [20]. This patient is presumed to have mutations in the genes regulating cell cycle progression as well as apoptosis in erythrocyte progenitors that are common in MDS, leading to anemia and ineffective erythropoiesis [21]. MDS bone marrow often shows reduced proliferation of BFU-E and increased accounts of apoptosis before erythrocyte maturation [3]. Specific chromosomal mutations have been identified and can be tied to specific instances in hematopoiesis where regulatory mechanisms are faulty. For instance, deletions of the chromosome arm 5q stabilize p53, increasing apoptosis before erythroid maturation, which is associated with a better prognosis, as these clonal populations do not grow uncontrollably [22]. Other variants of MDS include genetic mutations leading to defects in the mitochondrial control of iron [23]. |
| Further, it has been demonstrated that somatic mutations in hematopoietic progenitors accumulate with age, potentially leading to clonal abnormalities in hematopoietic cells, despite the absence of a diagnosis for a specific hematologic disease [24,25]. These conditions, referred to as age-related clonal hematopoiesis (ARCH) and clonal hematopoiesis of indeterminate potential (CHIP), are associated with an increased risk of developing hematological malignancies [24,25]. Similarly, premalignant conditions have been described in which patients present with either idiopathic cytopenia of undetermined significance (ICUS) or idiopathic bone marrow dysplasia of uncertain significance (IDUS) [26]. Patients with ICUS or IDUS may progress to develop a neoplastic disorder, although the presence of these conditions is not completely predictive for a subsequent malignancy [26]. In these cases, it is suggested that patients are treated similar to low-risk MDS until a further diagnosis is established [26]. |
| Since myelodysplastic syndromes are some of the most common hematologic malignancies in patients over 70, with an incidence rate exceeding 20 per 100,000 people per year [27], costeffective management strategies are needed. Low-risk MDS patients are often observed and treated with supportive care, such as transfusions [28]. The patient in this case report had a revised IPSS score of 2.5 indicating that he has low-risk MDS [29]. Patients with a higher risk disease are more commonly treated with DNA methyltransferase inhibitors [30] before being considered for hematopoietic stem cell transplantation (HSCT), the only treatment option for a potential cure [31]. Although fewer patients undergo a stem cell transplant than are eligible for it, approximately 30-40% of patients who receive HSCT become long-term survivors of MDS [1]. Monitoring this patient over time will help determine the progression of disease, and whether he requires a more aggressive treatment regimen, such as allogeneic HSCT. |
Figures at a glance
|
 |
| Figure 1 |
|
| |
| |

