Introduction
A common complication of water retaining diseases is hypervolemic hyponatremia, which is characterized by low blood sodium concentration (<135 mmol/L) and high circulating levels of arginine vasopressin (AVP) [1]. This AVP hypersecretion often results from insufficiency of efficient arterial volume such as in congestive heart failure (CHF) [2] and hepatic cirrhosis [3], and results in water accumulation in extracellular space in forms of edema or ascites. Under this condition, excessive fluid causes a “hypervolemia” but cannot form an efficient arterial volume, thereby creating a vicious circle between hypersecretion of AVP and the hyponatremia. The treatment of this hypervolemic hyponatremia is a strong challenge to the clinicians and the researchers. Targeting some known pathological mechanisms underlying this complication by blocking both V2 and V1a AVP receptors does improve the hyponatremic symptom; however, it does not reduce the morbidity and mortality [4]. For this reason, it is crucial to further explore mechanisms underlying hypervolemic hyponatremia and to identify reasons for the significant mortality.
AVP is a neuropeptide hormone mainly synthesized by magnocellular neurosecretory cells (MNCs) in the hypothalamus. MNCs containing AVP (i.e., AVP neurons) are identified in the supraoptic (SON) and paraventricular (PVN) nuclei as well as several accessory nuclei between the SON and PVN [5]. In these nuclei, another type of MNCs, oxytocin (OXT) neurons, coexists with AVP neurons and OXT can modulate AVP neuronal activity [6]. In these nuclei, the MNCs have close association with astrocytes structurally and functionally [7-8], and their interactions are an important factor determining AVP neuronal activity. In response to changes in blood volume, osmolality or pressure, the firing rate and pattern of AVP neurons also change correspondingly to determine the amount of AVP secretion from axonal terminals of AVP neurons in the posterior pituitary [9-10]. By increasing or decreasing AVP secretion, the AVP-secreting system can adjust water intake and salt appetite, functions of kidney, and the activity of endocrine and cardiovascular systems [11]. In this process, both systemic [12] and local [13] neurohumoral regulatory mechanisms are involved. The systemic regulation, involving several neurohumoral reflexes and blood-borne factors, is largely determined by efficient arterial volume or actual blood pressure, sympathetic nerve outflow, and the activity of renin-angiotensin (ANG)-aldosterone (ALD) system [14]. The local regulation is achieved by changing synaptic input [15], astrocytic-neuronal interactions [7-8], autoregulation of AVP neuronal activity [16-17], and sequential activation of specific cellular signaling molecules [18]. It has been proposed that in water-retaining diseases, nonosmotic stimuli override hyponatremic inhibition of AVP secretion [19], thereby removing inhibitory effects of hyponatremia on the activity of the local neural circuit. As a result, hypersecretion of AVP occurs in the presence of hyponatremia.
Here, we review the mechanisms underlying AVP hypersecretion in views of systemic neurohumoral control of AVP secretion, resetting osmosensory threshold at the local neural circuit, bimodal interactions between astrocytes and AVP neurons, and other cellular/molecular events (Table 1). In the text, we used hyponatremia and hypoosmolality exchangeably since most hyponatremic disorders are associated with hypoosmolality, and hypoosmotic stimulation is mainly based on low Na+ solution in the cited reports.
Osmotic regulation of AVP secretion
In the body, many specialized sensors can sense changes in the internal environment and trigger adaptive reactions to maintain hydromineral homeostasis. In neurohumoral modulation of AVP secretion, osmosensory neurons in the diencephalon, particularly those in the subfornical organ (SFO) [20], the antero-ventral third ventricular region (AV3V) [21], the organum vasculosum of the lamina terminalis (OVLT) and the SON [22], can sense changes in extracellular osmotic pressure and modulate AVP neuronal activity. In the SON, both AVP and OXT neurons are osmosensitive although they have different effects on hydromineral homeostasis [9]. In parallel with studying central osmosensors, peripheral osmosensory cells have also been identified, such as juxtaglomerular cells of the kidney [23], hepatic portal vein [24] and cardiomyocytes [12]; they also provide abundant information of osmotic regulation of AVP secretion.
In general, increases in extracellular osmolytes activate osmosensory cells while decreases in the osmolality reduce their firing activity [9]. In these osmosensory neurons, the osmosensitivity is due to the presence of intrinsic cellular mechanoreceptors and glial modulation. In neurons, osmosensing mechanoreceptors were initially identified as stretch-modulated cation channels [25] and now are associated to the transient receptor potential (TRP) vanilloid (V) family cation channel proteins. The osmosensory ability of SON neurons is also related to astrocytic sensitivity to osmotic changes, as known for the reactive release of gliotransmitters from astrocytes such as taurine, b-alanine [26] and D-serine [27] as well as the morphological plasticity of astrocytes [28]. In the following paragraphs, we will discuss the characteristics of osmosensory neurons and osmotic reflex, and leave astrocytic mediation of osmosensation in the section of Interactions between glia and AVP neurons.
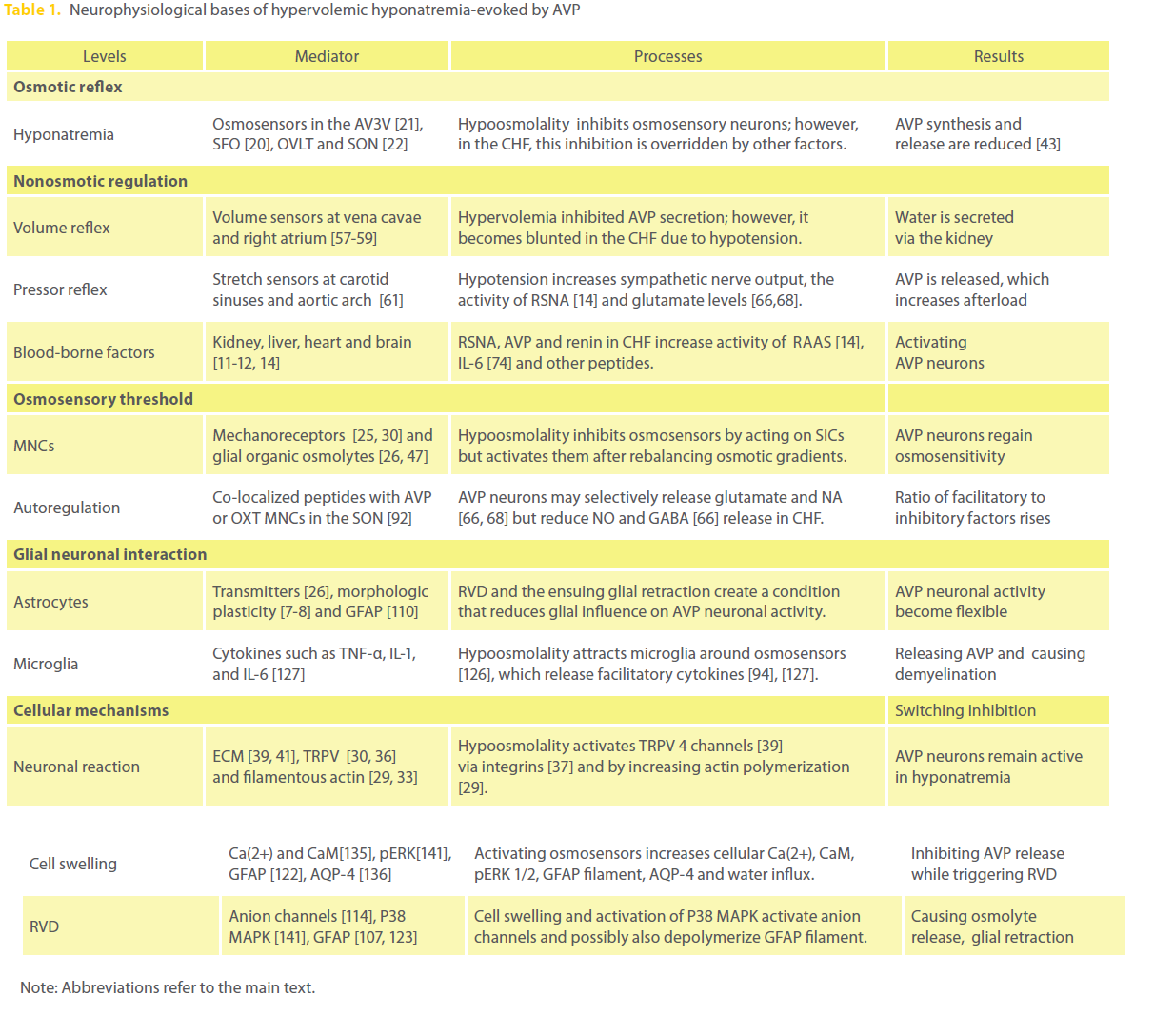
Table 1: Neurophysiological bases of hypervolemic hyponatremia-evoked by AVP
In the osmosensory neurons, the TRPV channels are sensitive to cell membrane stretch to permit the generation of a nonselective cation current, particularly for Ca2+ [29]. It is believed that osmotically-evoked activation of OVLT neurons is mediated by TRPV1 and TRPV4 channels; these OVLT neurons regulate the activity of downstream MNCs in the SON through graded changes in glutamate release [30]. This finding is confirmed by gene depletion studies that TRPV 1 [31] and TRPV 4 channels [32] are essential for hyperosmotic activation of osmosensory neurons. The activation of these TRPV channels is resulting from actin polymerization because hyperosmotic activation stimulated actin polymerization [33] and destruction of filamentous actin blocked hyperosmotic activation of osmosensory neurons [34]. While these results support the essential role of TRPV channels in sensation of hyperosmotic stimulation, observations have also revealed that the number of Fos-positive cells in the OVLT, the SON and PVN was similar between wild-type and TRPV1-/- mice after osmotic stimulation [35]. This finding suggests that individual TRPV channels may partially contribute to the osmotic reactions of osmosensory neurons and their deficiency can be compensated by other TRPV channels. Moreover, the interaction between TRPV4 and filamentous actin was involved in sensing hypoosmolality [29], and a mechanosensitive ion channel (gramicidin A) behaved either as a stretch-inactivated or as a stretch-activated channel depending on the lipid composition of the surrounding lipid bilayer [36]. Thus, TRPV channels are involved in the sensation of both hyper- and hypo-osmolality.
One question in studying these osmosensors is how the stretch message of osmotic challenges is delivered to the osmosensory neurons via these mechanoreceptors. Some hits to this question have emerged from studies on physiology of extracellular matrix (ECM). The fluid volume in the interstitial space is normally regulated within a narrow range by automatic re-adjustment of the interstitial hydrostatic and colloid osmotic pressures through capillary filtration and the lymphatics. During hyponatremia, more water gets into the interstitial tissues, which are taken up by b1-integrinassociated ECM [37]. In this process, integrins can be activated to trigger cellular reactions as identified in hepatocytes [24] and cardiomyocytes [38]. Moreover, both hyper- and hypoosmotic stimuli can activate TRPV4 channels by providing mechanical activation of integrins because mechanical forces applied to cell surface b1 integrins can rapidly activate Ca2+ influx through TRPV4 channels [39]. In the SON, polysialic acid-enriched neural cell adhesion molecule occurs on the surfaces of MNCs and astrocytes, and they can absorb large amounts of water to modulate cell adhesion [40]; expression of integrins-associated ECM organization has also been identified in the SON [41]. Thus, hypoosmotic challenges can directly activate TRPV channels in the osmosensory neurons through integrin signaling in the SON. Finally, relative to the pure facilitatory effect of hyperosmolality on AVP secretion, effects of hypoosmotic stimulation are biphasic. That is, short in vitro hypoosmotic stimulation evokes inhibition only [22, 42] while prolonged presence of hypoosmolality causes rebound increase in AVP secretion [42], an indicator of activation of AVP neurons. Thus, we propose that the activation of TRPV channels during hypoosmotic stimulation needs longer time than hyperosmotic stimulation does.
Changes in blood osmolality activate osmotic reflex, the major neurohumoral reflex in body hydromineral homeostasis, and modulate the activity of AVP neurons directly and indirectly via other osmosensors [9]. In superfused explants of rat hypothalamus, changes in firing rate and frequency of spontaneous EPSPs but not IPSPs of the MNCs are positively correlated to changes in external osmolality between 275 and 355 mOsm [22]. Brief (10-20 s) applications of hypoosmotic solutions to the OVLT caused prolonged (> 1 min) decreases in the firing activity of MNCs [22]. Moreover, AVP secretion was suppressed in healthy rat model of hypoosmolality that was produced using V2 AVP receptor agonist and a liquid diet [43]. Under the same condition, the somatic and nuclear sizes of AVP neurons decreased to approximately 60% of basal values; OXT and AVP mRNA levels reduced to approximately 10~20% of basal levels [44]. These findings indicate that short in vitro and chronic in vivo hypoosmotic stimuli are inhibitory for AVP neuronal activity and AVP secretion. On the other hand, prolonged in vitro hypoosmotic stimulation in a rate of 2% or 5% per hour over 2 h had a biphasic effect on AVP secretion: a short suppression (<60 min) followed by a rebound facilitation [42]. It was also found that AVP neurons in the SON cultures were significantly increased in 255 mOsm medium as compared to 300 mOsm medium [45]. These data suggest that different mechanisms are working for in vivo and in vitro hypoosmotic regulations and prolonged in vitro hypoosmotic actions may cause additional reactions of the brain osmotic regulatory system. Importantly, the rebound increase in AVP release during hypoosmotic stimulation in vitro matches those observed in hyponatremic patients with AVP hypersecretion [46]. This clue leads to our agreement with that in pathological hyponatremia of water retaining diseases, AVP hypersecretion is due to the presence of excitatory nonosmotic stimuli that override hypoosmolality-elicited inhibition of AVP neuronal activity as previously proposed [19,47] and/or due to activation of local facilitatory mechanisms.
AVP secretion is also related to peripheral osmosensors. Hepatic osmoreceptors, located in the region of the hepatic portal vein, are sensitive to changes in portal blood osmolality and can cause variations in plasma AVP and water excretion [9,48]. Thus, activation of the hepatic osmoreceptors can buffer major systemic osmotic changes. Macula densa cells in the kidney can detect changes in tubular fluid composition. Changes in luminal fluid NaCl concentration alter intracellular NaCl level and cell volume, which involves Na+/Cl- contransporter and a stretch-activated non-selective anion channel [49]. Macula densa signaling then changes the production and release of ATP, prostaglandin E2, nitric oxide (NO), and renin [49]. These paracrine signals can control juxtaglomerular function, renal vascular resistance [50] and alter AVP secretion by regulation of renin release [11]. In the heart, hyponatremia can also sensed by atrial cardiomyocytes. An osmotic stretch during hyponatremic stimulation evokes release of atrial natriuretic peptide (ANP) from swollen atrial cardiomyocytes [51]. ANP can suppress AVP secretion by directly inhibiting the activity of osmosensory neurons [52] and by antagonizing the facilitatory effects of angiotensin II (ANGII) on AVP neuronal activity [53- 54].
Nonosmotic modulation of AVP neuronal activity
In parallel with osmotic regulation of AVP secretion, cardiovascular activity, autonomic nerve activity, endocrine, metabolic activity, and personal habits like alcohol consumption and sleep/awake cycle can also modulate AVP secretion [55-56]. Among them, cardiovascular activity has bidirectional association with AVP neuronal activity and is the most important nonosmotic factor modulating AVP secretion, which involves blood volume and pressure as well as many blood-borne factors.
Changes in blood osmolality often change blood volume. For instance, dehydration can cause hypernatremia and hypovolemia simultaneously, which coordinately increase AVP release and the ensuing water retention [9]. Changes in blood volume naturally alter blood pressure and can change the activity of both low and high pressure-sensitive cardiovascular reflexes [11]. The atrial volume reflex is related to an expansion of blood volume in venous system and the activation of low pressure-sensitive reflex pathway. When hypervolemia occurs in the venous system, volume sensors at the vena cavae and right atrium become active and emit the volume-signals via vagal nerve to brain regions that are associated with the regulation of AVP secretion and sympathetic outflow [57]. In addition to inhibitory input from GABAergic neurons in the perinuclear zone of the SON [58], a group of neurons containing ANP in the the AV3V is also activated to inhibit AVP secretion and promote OXT release [59]. The reduction of AVP promotes water secretion while the increase in OXT level evokes ANP release from the heart [59]. ANP directly promotes Na+ and water excretion and thus, reduces water retention in the body and suppresses AVP secretion by brain actions [53-54]. Meanwhile, hypervolemic activation of vagal nerve reduces renal sympathetic nerve activity (RSNA) and cardiac output by suppressing the activity of parvocellular neurons in the PVN [60]. Thus, hypervolemia also increases blood perfusion of the kidney, which together with the decreased RSNA and AVP secretion reduces renin release and the subsequent production of ANG II and AVP release [11]. As blood AVP decreases, free water will be removed and blood volume is decreased. Likewise, hypovolemia causes opposite systemic regulatory reactions, leading to increases in AVP secretion and in blood volume [58-60].
Along with osmotic and atrial volume reflexes, changes in blood pressure also alter AVP secretion through baroreflex and pressor reflex. The stretch sensors or baroreceptors at the carotid sinuses and aortic arch are excited by hypertension, which reduces sympathetic nerve output but increases parasympathetic nerve output by acting on autonomic nuclei in the medulla. This fact has been well explored in the rabbit [61]. As a result, cardiac output and peripheral resistance are decreased, leading to a decrease in blood pressure. The reduction of sympathetic nerve output, particularly the RSNA, reduces the activity of RAAS [14] and indirectly decreases AVP secretion. On the contrary, hypotension can cause a pressor reflex via neurophysiological processes opposite from the baroreflex. In the pressor reflex, neuronal activity in the nucleus of the solitary tract (NST) and ventrolateral medulla (VLM) [62] as well as the osmosensory organs [63] are significantly activated. Hypotension-activated noradrenergic afferents from the NST and caudal VLM can activate AVP neurons [64]. Meanwhile, the GABAergic inhibitory input to AVP neurons from the diagonal band of Broca and the perinuclear zone of the SON are reduced [58,65]. The increases in the excitatory events and decreases in the inhibitory events strongly facilitate AVP secretion, which is particularly significant in the water-retaining diseases such as CHF. In patients with CHF, although an increased preload could trigger the atrial volume reflex, degeneration of myocardium and the enlargement of the heart volume can significantly reduce the sensitivity of volume sensors to an expansion of blood volume, thereby reducing volume reflex. At the same time, stretch-elicited ANP release is also reduced, which weakens the antagonistic effect of ANP on ANGII-elicited AVP secretion [53-54]. To worsen this situation, the reduction of cardiac output decreases the efficient arterial volume and causes hypotension, which triggers a strong pressor reflex, as hepatic cirrhosis does [3]. Thus, a potential atrial volume reflex is masked by the pressor reflex [62]. The resultant activation of sympathetic outflow and RAAS as well as increases in AVP secretion and peripheral resistance could partially restore the blood pressure. However, these changes do not repair the damaged cardiac tissues but increase their afterload, which could further damage the degenerated cardiac tissues and reduce the efficient arterial volume. Under this condition, the RAAS remains active due to the reduction in kidney perfusion and activation of RSNA [11, 14]. The activation of RAAS together with other blood-borne factors and direct neural activation strongly increase AVP release.
In reviewing interactions between these nonosmotic and osmotic factors, one crucial question is: what accounts for the failure of hyponatremia to exert its suppressive effects on AVP secretion at the local neural circuit? It has been reported that in the PVN of rats with heart failure, there are higher levels of glutamate, noradrenalin (NA) and ANG I receptor, and lower levels of GABA, neuronal NO synthase (nNOS), and the 67- kDa isoform of glutamate decarboxylase (GAD67, the enzyme converting glutamate to GABA) [66]. In hepatic encephalopathy, expression of astrocyte glutamate transporter GLT-1 is also decreased, resulting in reduction of glutamate transport and thus an enhancement of extracellular glutamate levels [67-68]. While these excitatory nonosmotic influences on AVP neurons blunt atrial volume reflex in heart failure [69], they could also negate or neutralize the inhibitory effects of hyponatremia on AVP secretion in view of the neurohumoral regulation that hypoosmotic inhibition is largely derived from suppression of excitatory input to AVP neurons [22].
In the generation of hypersecretion of AVP, blood-borne factors, particularly peptide hormones, play critical roles. By acting on osmosensory brain areas where blood brain barrier (BBB) is not intact, these peptides can access osmosensory neurons and directly change their activity, leading to changes in AVP secretion. Besides the role of RAAS discussed above, other peptides implicated in the activation of AVP neurons are endothelin [70], relaxin [71], L-thyroxin [72], secretin [73], interleukin-6 [74], and others such as kisspeptin-10 [75] and neuronostatin [76]. It is particularly interesting to examine the roles of AVP in the generation of hyponatremia. Under pathological conditions, excessive activation of AVP receptors can worsen the hyponatremia and cause brain damage. It is generally accepted that AVP actions are mediated by three AVP receptors: V1a, V1b and V2 [5]. V2 receptor activation promotes water reabsorption in renal medulla by increasing the expression and function of a water channel protein, aquaporin (AQP)-2 [77] and Na-K-2Cl co-transporter [78]. These effects promote water and sodium reabsorption and form a basis of hyponatremia. V1a receptor is mainly located at vascular smooth muscles. The activation of V1a receptor can activate RAAS and sympathetic nerve outflow [79]. Activation of this receptor can also stimulate Na+/H+ exchanger activity in the BBB [80] and thus, promote brain edema and infarction [81]. V1b receptor is not required for normal MNC function; however, overactivation of V1b receptor can worsen brain edema by modulating astrocytic water permeability [82]. A sustained stimulation of AVP receptors has been found to impair cerebral vasculature [83] and kidney functions [84]. Taken together, increases in AVP secretion can promote water retention and vasoconstriction whereas excessive activation of AVP receptors could exacerbate brain edema during hyponatremia, even brain and kidney damage, leading to further hydromineral imbalance and worsening of the primary diseases.
Resetting osmosensory threshold at the local neural circuit
In the osmotic regulation, AVP secretion is under the modulation of local neural circuits involving synaptic input, interactions between astrocytes and AVP neurons, and autoregulation of AVP neuronal activity, etc. The synaptic input is the essential link of systemic regulation of AVP neuronal activity while other local factors constitute an inherent facilitatory machinery of AVP secretion during prolonged hyponatremia. In rat hypothalamic explants, hypoosmotic stimulation induced prolonged AVP release following a brief inhibition [42]. This biphasic feature of AVP secretion during hyponatremia indicates a reduction of osmosensory threshold in osmosensors or an activation of inherent facilitatory machinery of AVP neuronal activity. This proposal is supported by a number of other observations. For instance, a reduction of hyperosmotic stimulation triggered a rebound increase in AVP release after a short decrease in the hypothalamic explants [42]; low plasma osmolality was sensed as “normal” by AVP neurons in pregnant rats [85]; direct osmotic stimulation of the SON and PVN by microdialyzing hyperosmotic medium increased intranuclear release of AVP, which was further enhanced after replacement of hyperosmotic with isoosmotic fluid [86]; and the expression of β-adrenoceptors in astrocytes, a sensor of NA, can be re-boosted during the rehydration in dehydrated rats [87]. Obviously, a relative reduction of osmolality can increase AVP neuronal sensitivity to osmotic and nonosmotic stimuli at the level of local neural circuits.
Several lines of neurophysiological processes can explain this phenomenon including adaptive features of AVP neurons, changes in the neurochemical environment and glial neuronal interactions. Here, we will examine the first two features. First, the reduction of osmosensory threshold is attributable to an adaptive change in the osmotic sensitivity of AVP neurons. When decreased osmolality leads to cell swelling, a stretchinactivated cation channel (SIC) of osmosensory neurons in the hypothalamus is inactivated, which hyperpolarizes the MNCs and reduces sensitivity of the MNCs to the excitatory input [63]. Theoretically, the SICs could become sensitive to osmotic change again when the “excessive water” is evenly distributed in whole cells (away from the membrane) or water efflux occurs due to relative increases in extracellular osmolality. Actually, in the cell volume regulation, organic osmolytes are released following the swelling of astrocytes [47], which could activate AVP neurons through the activation of SICs [30]. In addition, the secondary activation of MNCs can also be mediated by integrin-mediated activation of TRPV channels [37, 39]. It will also be very interesting to examine the contribution of newlyidentified melastatin-related subfamily of TRP channels, TRPM4 and TRPM5 [88] to the adaptation of osmosensory neurons.
At the local neural circuit, osmotic modulation of autoregulatory process of AVP neurons could also change the balance between excitatory and inhibitory neurotransmitters and neuropeptides, which may contribute to this hypersensitivity of AVP neurons. NO is a negative feedback mediator of ANG II-elicited AVP secretion [89], partially via increasing GABA release [90]. The reduction of NO production and GABA levels in the PVN in heart failure [66] could facilitate AVP secretion elicited by glutamatergic transmission [91]. In addition, many co-localized peptides in AVP neurons [92] could be involved in the reduction of osmosensory threshold. Under pathological conditions, facilitatory peptides for AVP secretion can be released within the brain while inhibitory peptides remain unchanged or reduced. For instance, when AVP release is increased, inhibitory apelin accumulates in these neurons rather than being released [93]. In elderly people, the level of facilitatory interleukin-6 (IL-6) is high while inhibitory insulin growth factor-I (IGF-I) is low [94], which partially accounts for the high morbidity of elderly patients with hyponatremia [1]. As a whole, adaptive changes in osmosensors and increases in local facilitatory factors for AVP neuronal activity as well as special glial AVP neuronal interactions (see below) cause a reduction of osmosensory threshold and make AVP neurons more sensitive to systemic nonosmotic stimuli.
Interactions between glia and AVP neurons
It has been shown that acute hyperosmolality-induced Fos expression in the SON depends on normal functions of astrocytes [95]; thus, the participation of astrocytes is an essential step in systemic regulation of AVP neuronal activity. The interactions between astrocytes and AVP neurons are multiple. (1) Astrocytes mediate the communication and volume exchanges between blood vessels and neurons through rich vascular networks [96] and AQP-4 on astrocytes around the MNCs [97]. (2) Astrocytes can sense changes in neuronal activity by expressing neurotransmitter and/or neuromodulator receptors such as adrenergic [98], adenosine A2A [99], OXT [100], and GABA receptors [101]. (3) Astrocytes are a rich source of neurochemicals such as taurine [102], glutamate [103], and D-serine [27], and can release these gliotransmitters in response to changes in local neurochemical environment. (4) Astrocytes can buffer changes in extracellular environment such as water through AQP-4 [97], K+ though SK3 Ca2+-dependent K+ channel [104], GABA by GABA transporter 3 [105] and glutamate via GLT-1 [106]. (5) There are metabolic couplings between astrocytes and MNCs. For instance, astrocytes provide glutamine to the MNCs, which is converted from glutamate by glutamine synthetase [107]. (6) The activity of AVP neurons can change astrocytic plasticity. For instance, brain AVP causes astrocyte swelling by activating both V1a [83] and V1b [82] receptors, and active neuronal activity in the SON increases intracellular Ca2+ levels in astrocytes by ATP and NA [98]. Meanwhile, AVP neurons can also sense gliotransmitters and change their firing activity echoing astrocytic plasticity such as inhibitory responses to astrocytic taurine [102], excitatory responses to D-serine [27]. (7) There are rich gap junctions between astrocytes in the SON, PVN and the posterior pituitary [108] and between neurons and astrocytes in the SON [109]. The extensive junctional connections make astrocytes and AVP neurons a functional syncytium. By this syncytium, astrocytic modulation of AVP neuronal activity could be greater than any single source of synaptic input in response to systemic challenges. (8) Morphological plasticity of astrocytes determines the strength of interactions between astrocytes and AVP neurons. A retraction of astrocytic processes around AVP neurons increases their relative distance and then reduces astrocytic influences of AVP neuronal activity [7-8]. As a result, cellular apposition and junctional coupling between AVP neurons increase significantly, which promotes mutual excitation of neighboring AVP neurons [7]. Finally, plastic changes in glial fibrillary acidic protein (GFAP), a major cytoskeletal element of astrocytes, largely determine the morphological and functional plasticity of astrocytes through direct molecular interactions with AQP-4, glutamine synthetase and actin filaments and others [110]. These structural and functional features determine a critical position of astrocytic plasticity in systemic and local modulation of AVP neuronal activity.
During hypoosmotic challenges, interactions between astrocytes and AVP neurons turn an initial inhibitory action into a facilitatory effect. When blood osmolality decreases, excessive water can get into the brain through BBB, which creates a hypoosmotic environment in the brain [80]. In this environment, a bimodal cell volume regulation occurs: swelling and the ensuing regulatory volume decreases (RVD) in both astrocytes [26] and neurons [111]. Relative to neurons, astrocytes are more sensitive to the osmotic change due to their high ability to take up excessive water and ions rapidly through AQP-4 and associated ionic channels [104, 112] while volume modulation in the MNCs is relative weak [113]. It has been observed in vitro that hypoosmotic stimulation caused astrocyte swelling within minutes, followed by a compensatory volume-dependant release of organic osmolytes and osmotically-obligated water [47]. The cell swelling activates several anion channels, particularly the volume-activated anion channels and the volume-sensitive outwardly rectifying anion channel [114]. Their activation leads to extrusion of intracellular osmolytes such as K+, Cl- and small organic molecules including taurine, glutamate, and D-aspartate and β-alanine, and results in the RVD [115]. By transferring excessive astrocytic volume into extracellular space and finally into blood vessels in the hypothalamus [96-97], the RVD of astrocytes could attenuate neuronal swelling and maintain MNC functions. It is worth noting that in the RVD, Na+-K+-ATPase plays an important role by removing Na+ from the cells [116]. The severity of hyponatremia in menstruant women has been attributed to an inhibitory effect of estrogen on Na+-K+-ATPase and the ensuing long cell swelling [116]. In patient of water-retaining diseases, the encephalopathy can occur either acutely in 24-48 hours or in several days [47]. This is clearly due to the huge volume of the brain and the buffer ability of whole body compared to the cell culture of hypothalamic explants. However, a consequence of brain swelling among the survivals is brain volume reduction from single cells to whole brain [47], which matches the in vitro effects of hypoosmolality on astrocytic activity. The general match credits the usage of in vitro hypoosmotic study in revealing hyponatremic neuropathogenesis despite their temporal differences.
Mechanistically, the initial cell swelling is consistent with a dramatic inhibition of AVP neuronal activity [22] and the reduced AVP secretion [43] . This is related to the release of inhibitory astrocytic taurine and β-alanine at early stage of hyponatremia [115]. Taurine is an endogenous agonist of glycine receptor [117]; β-alanine is the inhibitor of GABA transporter and can increase extracellular GABA levels [118]. Thus, both taurine and β-alanine can inhibit AVP neuronal activity following their release from astrocytes during the RVD. In addition, by extending astrocytic processes around AVP neurons during cell swelling, hyponatremia can also inhibit AVP neuronal activity by reducing extracellular K+, glutamate and interneuronal interactions that normally facilitate AVP secretion as previously reviewed [7-8].
Mechanisms underlying the later rebound increase in AVP release [42] or brain volume reduction [47] is more complex relative to the initial inhibition. The rebound increase in AVP secretion cannot be explained merely by a conversion of inhibitory to excitatory influences of hyponatremia on AVP neurons directly or by acting on their presynaptic excitatory input, since a pure hyponatremic condition caused only inhibitory responses of AVP neurons in vivo [30]. Alternatively, astrocytic RVD can account for the increased AVP secretion by the following approaches. (1) The release of organic osmolytes during the RVD [26] could form a relatively high osmotic microenvironment around AVP neurons. The relative high osmolality in turn causes shrinkage of AVP neurons by drawing water out of the MNCs, and then activates the SICs [30] to allow more AVP release. (2) Losing contents in the astrocytes during RVD [26] could cause a retraction of astrocytic processes around AVP neurons, which decreases the inhibitory influence of astrocytes on AVP neurons [7-8]. (3) Glia retraction can dramatically reduce gap junctional interactions, particularly those between astrocytes and AVP neurons. The reduction of junctional communication frees AVP neurons from synergistic restriction or influence of astrocytic activity on neuronal activity and thus, makes the response of individual AVP neurons more flexible to systemic stimuli. (4) Loss of the inhibitory taurine and β-alanine following the RVD [26, 119] also contributes to the high excitability of AVP neurons during prolonged hyponatremia. Obviously, these factors can contribute to the reduced osmosensory threshold in AVP neurons.
In the generation of cell swelling and the ensuing RVD, GFAP could be an important contributor. GFAP plasticity largely determines the extension and retraction of astrocytic processes [120] in association with changes in AQP-4 expression and actin filament in the SON [107]. Thus, increases in GFAP expression could account for cell swelling. This proposal is supported by the findings that astrocyte swelling evoked by high K+ and hypoosmotic solution is significantly slower in GFAP-deficient mice compared to wild-type mice [121] and that loss of GFAP resulted in an inhibition of hypertrophy of astrocytic processes following hypoosmotic stimulation [122]. Likewise, a reduction of GFAP could facilitate the RVD by increasing its association with actin filament to cause the retraction of plasma membrane and by decreasing its association with AQP-4 to reduce water influx as previously discussed [110]. Moreover, patients of hepatic encephalopathy is associated with a loss of GFAP [123] or no increase in GFAP [124], suggesting that GFAP is reduced after the initial increase during cell swelling [121-122]. Thus, the RVD and GFAP-guided glial retraction are functionally synergistic, and could jointly facilitate AVP neuronal activity.
In addition to astrocytes, microglial cells are also implicated in neurophysiological regulation of AVP neuronal activity [125]. Microglia are known to repair damaged tissue and control infection by phagocytosing apoptotic and necrotic cells; their activation can also increase AVP secretion. Local hyponatremia and cell swelling can increase chloride influx in microglia and promote the formation of lamellipodia [126]. Since lamellipodium extension and retraction are the driving force for cell migration, hyponatremia could attract microglia into brain tissues. Since osmosensors around the AV3V are not shielded by BBB, they should be the most accessible region for microglia to move in and could promptly receive the influence of microglial cytokines when hyponatremia occurs. Microglia are a rich source of cytokines including tumor necrosis factor (TNF-α), IL-1, and IL-6 [127], etc. These cytokines can either directly increase AVP secretion such as IL-6 [94] or indirectly by increasing IL-6 [127]. Thus, the activation of microglia can promote AVP hypersecretion.
Cellular mechanisms underlying AVP hypersecretion
Cellular mechanisms directly determine responses of AVP neurons to systemic and local stimuli. The cellular mechanisms include direct reactions of AVP neurons to osmotic stimuli and their responses to changes in the neurochemical environment. In response to osmotic challenges, a dilution of ions and organic osmolytes occurs in extracellular space around AVP neurons. The low extracellular Na+ reduces the driving force for action potential generation and in turn inhibits AVP neuronal activity [30]. The low Na+ inhibition could be alleviated by membrane depolarization due to a potential intracellular Na+ accumulation, resulting from an inhibition of Na+/K+ pump by lower extracellular K+ and AVP-elicited hypoxia [128]. Long term hypoosmotic influence is also related to changes in genomic processes and cellular structures [129], which remains to be further studied in CHF model. Consensus is that cell swelling and the RVD, particularly in astrocytes, are critical in the hypersecretion of AVP. This volume regulation involves a series of cellular and molecular events such as ECM [130], receptors [87, 131], actin cytoskeleton [33], ionic channels [56], etc. These events occur in certain spatiotemporal orders under the guidance of sequentially activated signaling molecules that are elicited by hyponatremia.
The first hypoosmotic signal triggering cell swelling is Ca2+. It has been reported that decreases in extracellular osmolality or ionic strength initially cause mild cell swelling, which activates Ca2+-permeable cation channels such as TRPV 4 [132] and leads to a net influx of Ca2+ from the extracellular space. This Ca2+ influx and/or the increase in membrane tension further trigger Ca2+ release from intracellular stores [133]. The resultant increase in intracellular Ca2+ levels and subsequent activation of calmodulin (CaM) kinase [134-135] are essential for cell swelling and the ensuing RVD. As shown in rat cerebral astrocytes, intracellular application of monoclonal anti-CaM antibody blocked hypoosmolality-activated volumeregulated anion channels [135]. Ca2+ entry and the activation of CaM increase membrane expression of AQP-4 [136], which facilitates clearance of extracellular K+ and glutamate and moves free water into the astrocytes [112, 137]. Another result of Ca2+ increase in swollen astrocytes is the activation of NOS. Activation of NOS has a dual effect on AVP neuronal activity: inhibiting AVP neurons by NO-related GABA release [90] and damaging multiple functional molecules by protein tyrosine nitration (PTN), particularly those metabolizing glutamate [134]. The inhibitory effect of NO is a transient event and could be removed as the decrease in brain NO production in heart failure [66] whereas, PTN-mediated excitatory effect can exert a long lasting effect even if NO production is reduced as discussed below.
In the pathogenesis of brain edema, increases in glutamate around MNCs [66, 68] create a self-amplifying signaling loop by activation of NMDA receptors and the ensuing PTN, initiated by the hypoosmotic stimulation, particularly in those accompanying with hyperammonemia in hepatic cirrhosis [138]. An overactivation of NMDA receptor, mainly at the extrasynaptic sites due to GLT-1 inhibition [68], increases NO production and the ensuing PTN formation [139]. The PTN was colocalized with GFAP and glutamine synthetase [140] as well as glyceraldehyde 3-phosphate dehydrogenase (GAPDH), extracellular signal-regulated protein kinase (ERK) 1/2 and the peripheral benzodiazepine receptor (an allosteric binding site of GABAA receptors) [139]. Among them, nitrosylation of glutamine synthetase has been associated to inactivation of this enzyme [139]. The inactivation of glutamine synthetase, inhibition of GLT-1 [68] and reduction of GAD67 [66] collectively increase extracellular glutamate levels, and facilitate AVP secretion. In addition, hypersecretion of AVP under pathological hyponatremia could also cause malfunction of GFAP plasticity, energy metabolism, GABAergic action, and cell signaling due to a potential damaging effect of PTN on GFAP, GAPDH, and benzodiazepine receptor. If the hyponatremic and/or hyperammonic states are not resolved promptly, the damage to normal cellular/molecular regulation could be irreversible. Thus, blocking the formation or relieving the actions of PTN will be the key for restoration of the cellular processes controlling AVP secretion.
Downstream to the Ca2+/CaM, phosphorylated (p) ERK 1/2 could be a crucial signal in the cell volume regulation. Following Ca2+ entry, pERK 1/2 is increased as shown in trout hepatocytes [141]. In this preparation, chelating extracellular Ca2+ to prevent Ca2+-associated cell swelling also abolished hypoosmolalityelicited pERK 1/2 increases. The following evidence supports a functional role of pERK 1/2 in cell volume regulation. Blocking the activation of ERK 1/2 [141] significantly reduced cell swelling and the ensuing RVD; GFAP expression depends on increases in pERK 1/2 [142]. Thus, pERK 1/2 is the downstream event of Ca2+/CaM signals and plays a critical signaling role in the volume regulation. Other signaling molecules involved in hypoosmotic regulation are Src kinase [38], p38 mitogenactivated protein kinase P38 MAPK [141], protein kinase C [143] and phosphoinositide 3-kinase [144], etc. Among these molecules, has been identified as another key signal for the occurrence of RVD. P38 MAPK was increased following the increases in pERK 1/2 and remained at high levels even when pERK 1/2 decreased, and blocking this kinase also blocked the RVD in an efficiency more than blocking ERK 1/2 [141]. Thus, it is possible that activation of ERK 1/2 is responsible for GFAP polymerization and cell swelling as well as the initiation of the RVD while activation of P38 MAPK is the major player of hypoosmolality-elicited RVD.
Taken together, the cellular mechanisms underlying AVP hypersecretion is as the follow. In response to hypoosmotic stimulation under the pathological conditions, strong NMDA receptor activation and membrane tension activate Ca2+ permeable cationic channels, and that triggers intracellular Ca2+ mobilization. Increases in intracellular Ca2+ levels activate CaM kinase, NOS and pERK 1/2. These factors together with expansion of GFAP and AQP-4 expression promote cell swelling. A subsequent activation of P38 MAPK and GFAP collapse together with activation of anion channels result in the RVD. RVD and the PTN create an excitatory environment around AVP neurons, thereby leading to AVP hypersecretion upon the prevailing actions of nonosmotic stimuli over the inhibitory hypoosmotic reflex. This proposal is, in general, consistent with hypoosmotic regulation of cellular volume in hepatic encephalopathy [145]. Figure 1 summarizes the hypothetic neurophysiological mechanisms underlying AVP secretion.
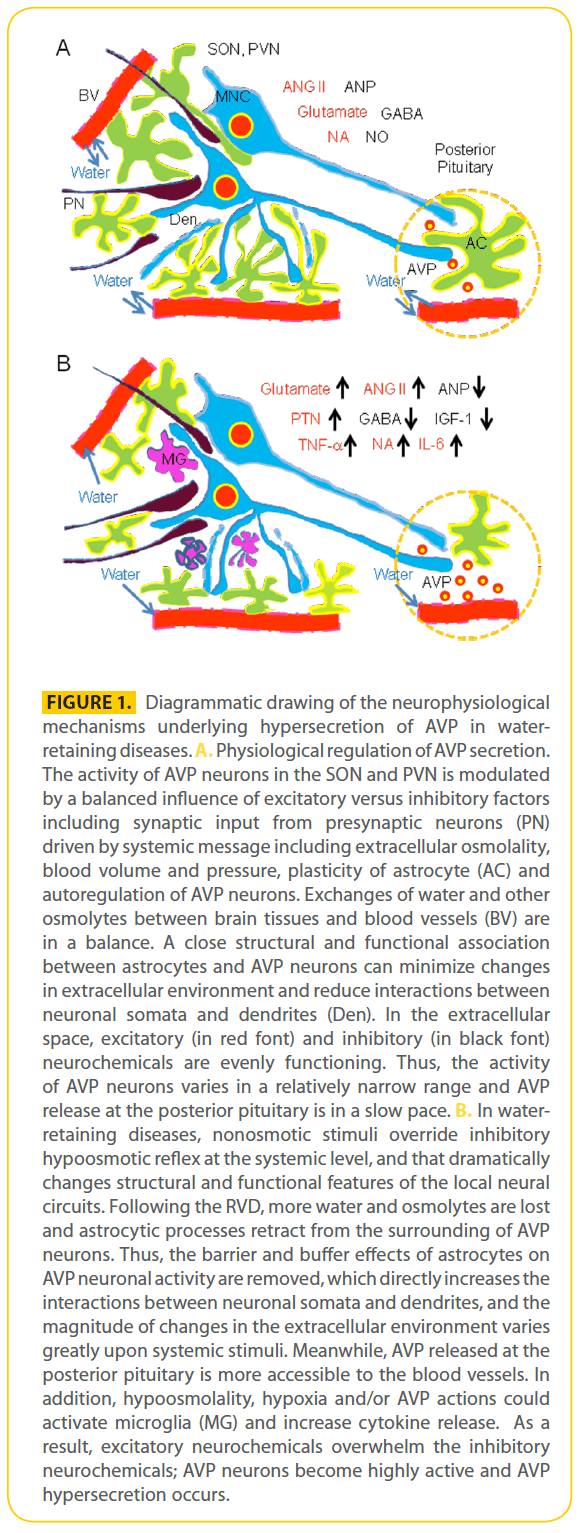
Figure 1: Diagrammatic drawing of the neurophysiological mechanisms underlying hypersecretion of AVP in waterretaining diseases. A. Physiological regulation of AVP secretion. The activity of AVP neurons in the SON and PVN is modulated by a balanced influence of excitatory versus inhibitory factors including synaptic input from presynaptic neurons (PN) driven by systemic message including extracellular osmolality, blood volume and pressure, plasticity of astrocyte (AC) and autoregulation of AVP neurons. Exchanges of water and other osmolytes between brain tissues and blood vessels (BV) are in a balance. A close structural and functional association between astrocytes and AVP neurons can minimize changes in extracellular environment and reduce interactions between neuronal somata and dendrites (Den). In the extracellular space, excitatory (in red font) and inhibitory (in black font) neurochemicals are evenly functioning. Thus, the activity of AVP neurons varies in a relatively narrow range and AVP release at the posterior pituitary is in a slow pace. B. In waterretaining diseases, nonosmotic stimuli override inhibitory hypoosmotic reflex at the systemic level, and that dramatically changes structural and functional features of the local neural circuits. Following the RVD, more water and osmolytes are lost and astrocytic processes retract from the surrounding of AVP neurons. Thus, the barrier and buffer effects of astrocytes on AVP neuronal activity are removed, which directly increases the interactions between neuronal somata and dendrites, and the magnitude of changes in the extracellular environment varies greatly upon systemic stimuli. Meanwhile, AVP released at the posterior pituitary is more accessible to the blood vessels. In addition, hypoosmolality, hypoxia and/or AVP actions could activate microglia (MG) and increase cytokine release. As a result, excitatory neurochemicals overwhelm the inhibitory neurochemicals; AVP neurons become highly active and AVP hypersecretion occurs.
Concluding remarks
Understanding the neurophysiological bases of hypervolemic hyponatremia-evoked by hypersecretion of AVP is a critical step to control water-retaining diseases and to extend the life-span of these patients. In clinical control of hypervolemic hyponatremia, common measures include prevention of volume overload, neurohormonal blockade, preservation of renal function, restraint of fluid intake, hyperosmotic saline infusion, and discontinuing drugs that may worsen hyponatremia, etc [1,4]. However, even using vaptans to block both V1a and V2 receptors, the most popular modern therapy of the hyponatremia, cannot reduce the morbidity and mortality in CHF [146]. Reviewing reasons that can account for the mortality reveals that high circulating AVP level is nevertheless an important factor due to its facilitation of brain edema and damaging effect on the BBB. The possible involvement of AVP itself can be foreseen from the preventive effect of re-introduction of hyponatremia on the demyelination, a common complication that occurs during hyperosmotic correction of the hyponatremia. The occurrence of demyelination is related to increases in BBB permeability and microglial activation [147]. Interestingly, re-induction of hyponatremia but not reduction of BBB permeability only resulted in a significant decrease in mortality after the correction of chronic hyponatremia in rats [148]. Hyponatremia can inhibit AVP secretion, at least in a short time frame [129]; thus, the effect of re-introduction of hyponatremia could be due to an inhibition of AVP secretion. Moreover, minocycline, an inhibitor of microglial activation, also decreases BBB permeability, decreases the expression of IL-1 and PTN, and reduces the loss of GFAP immunoreactivity [149]. All these microglial factors or actions are related to AVP hypersecretion during hyponatremia; thus, it further strengths the possibility that a reduction of AVP levels is involved in the therapeutic effect. In support of this possibility, lovastatin, an agent that can antagonize vasoconstriction effects of AVP [150], was also found to reduce the demyelinative lesions by decreasing the accumulation of microglia and their expression of TNF-α [147]. Thus, the possibility is high that the hypersecreted AVP plays a pivotal role in the generation of demyelination. For this reason, even though vaptans and other measures can correct the hyponatremic symptoms but they could not repair the damaged brain tissues or block some unknown harmful effects of the hypersecreted AVP, thereby failing to reduce the mortality.
Therapeutically efficient approach of hyponatremia by inhibition of AVP hypersecretion, however, would not be established until more direct evidence is available. Therefore, further exploration of the neurophysiological bases underlying hypervolemic hyponatremia will undoubtedly enlighten our strategies to control the hypervolemic hyponatremia and reduce the mortality of water-retaining diseases.
Acknowledgement
This work is supported by intramural fund of LSU Health Sciences Center-Shreveport. *These authors contributed equally to this work.
2540
References
- Shea AM, Curtis LH, Szczech LA, Schulman KA (2008) Sensitivity of International Classification of Diseases codes for hyponatremia among commercially insured outpatients in the United States. BMC Nephrol, 9:5.
- Jao GT, Chiong JR (2010) Hyponatremia in acute decompensated heart failure: mechanisms, prognosis, and treatment options. Clin Cardiol 33:666-671.
- Cordoba J, Garcia-Martinez R, Simon-Talero M (2010) Hyponatremic and hepatic encephalopathies: similarities, differences and coexistence. Metab Brain Dis 25:73-80.
- Schrier RW, Masoumi A, Elhassan E (2009) Role of vasopressin and vasopressin receptor antagonists in type I cardiorenal syndrome. Blood Purif 27:28-32.
- Burbach JP, Luckman SM, Murphy D, Gainer H (2001) Gene regulation in the magnocellular hypothalamo-neurohypophysial system. Physiol Rev 81:1197-1267.
- Hatton GI, Wang YF (2008) Neural mechanisms underlying the milk ejection burst and reflex. Prog Brain Res 170:155-166.
- Hatton GI (2004) Dynamic neuronal-glial interactions: an overview 20 years later. Peptides 25:403-411.
- Theodosis DT, Poulain DA, Oliet SH (2008) Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev 88:983-1008.
- Bourque CW, Oliet SH, Richard D (1994) Osmoreceptors, osmoreception, and osmoregulation. Front Neuroendocrinol 15:231-274.
- Gainer H, Sarne Y, Brownstein MJ (1997) Biosynthesis and axonal transport of rat neurohypophysial proteins and peptides. J Cell Biol 73:366-381.
- Thornton SN (2010) Thirst and hydration: physiology and consequences of dysfunction. Physiol Behav 100:15-21.
- Grindstaff RR, Cunningham JT (2001) Cardiovascular regulation of vasopressin neurons in the supraoptic nucleus. Exp Neurol 171:219-226.
- Iremonger KJ, Bains JS (2008) Dynamic synapses in the hypothalamic-neurohypophyseal system. Prog Brain Res 170:119- 128.
- Thomas GD (2011) Neural control of the circulation. Adv Physiol Educ 35:28-32.
- Sladek JR, Jr., Sladek CD (1985) Neurological control of vasopressin release. Fed Proc 44:66-71.
- McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, et al. (2003) The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol 35:901-918.
- Brown CH, Scott V, Ludwig M, Leng G, Bourque CW (2007) Somatodendritic dynorphin release: orchestrating activity patterns of vasopressin neurons. Biochem Soc Trans 35:1236- 1242.
- Tobin VA, Ludwig M (2007) The actin filament and dendritic peptide release. Biochem Soc Trans 35:1243-1246.
- Verbalis JG (1993) Osmotic inhibition of neurohypophysial secretion. Ann N Y Acad Sci 689:146-160.
- Matsumura K, Simon E (1990) Increase in basal firing rate and sensitivity to angiotensin II in subfornical organ neurones of ducks adapted to salt water. J Physiol 429:297-308.
- Honda K, Negoro H, Higuchi T, Tadokoro Y (1989) The role of the anteroventral 3rd ventricle area in the osmotic control of paraventricular neurosecretory cells. Exp Brain Res 76:497-502.
- Richard D, Bourque CW (1995) Synaptic control of rat supraoptic neurones during osmotic stimulation of the organum vasculosum lamina terminalis in vitro. J Physiol 489 ( Pt 2):567- 577.
- Baumbach L (1980) Renin release from isolated rat glomeruli: effects of colchicine, vinca alkaloids, dimethylsulphoxide, and cytochalasins. J Physiol 299:145-155.
- Haussinger D (2008) Osmosensing and osmosignaling in the liver. Wien Med Wochenschr 158:549-552.
- Voisin DL, Bourque CW (2002) Integration of sodium and osmosensory signals in vasopressin neurons. Trends Neurosci 25:199-205.
- Pasantes-Morales H, Alavez S, Sanchez Olea R, Moran J (1993) Contribution of organic and inorganic osmolytes to volume regulation in rat brain cells in culture. Neurochem Res 18:445- 452.
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, et al. (2006) Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125:775-784.
- Hatton GI, Perlmutter LS, Salm AK, Tweedle CD (1994) Dynamic neuronal-glial interactions in hypothalamus and pituitary: implications for control of hormone synthesis and release. Peptides 5 Suppl 1:121-138.
- Becker D, Bereiter-Hahn J, Jendrach M (2009) Functional interaction of the cation channel transient receptor potential vanilloid 4 (TRPV4) and actin in volume regulation. Eur J Cell Biol 88:141-152.
- Bourque CW, Ciura S, Trudel E, Stachniak TJ, Sharif-Naeini R (2007) Neurophysiological characterization of mammalian osmosensitive neurones. Exp Physiol 92:499-505.
- Yokoyama T, Saito T, Ohbuchi T, Hashimoto H, Suzuki H, et al (2010) TRPV1 gene deficiency attenuates miniature EPSC potentiation induced by mannitol and angiotensin II in supraoptic magnocellular neurons. J Neurosci 30:876-884.
- Liedtke W, Friedman JM (2003) Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci US A 100:13698-13703.
- Prager-Khoutorsky M, Bourque CW (2010) Osmosensation in vasopressin neurons: changing actin density to optimize function. Trends Neurosci 33:76-83.
- Zhang Z, Bourque CW (2008) Amplification of transducer gain by angiotensin II-mediated enhancement of cortical actin density in osmosensory neurons. J Neurosci 28:9536-9544.
- Taylor AC, McCarthy JJ, Stocker SD (2008) Mice lacking the transient receptor vanilloid potential 1 channel display normal thirst responses and central Fos activation to hypernatremia. Am J Physiol Regul Integr Comp Physiol 294:R1285-1293.
- Kung C, Martinac B, Sukharev S (2010) Mechanosensitive channels in microbes. Annu Rev Microbiol 64:313-329.
- Reed RK, Rubin K (2010) Transcapillary exchange: role and importance of the interstitial fluid pressure and the extracellular matrix. Cardiovasc Res 87:211-217.
- Ren Z, Raucci FJ, Jr., Browe DM, Baumgarten CM (2008) Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res 77:73-80.
- Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, et al. (2010) Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb) 2:435-442.
- Theodosis DT, Piet R, Poulain DA, Oliet SH (2004) Neuronal, glial and synaptic remodeling in the adult hypothalamus: functional consequences and role of cell surface and extracellular matrix adhesion molecules. Neurochem Int 45:491-501.
- Qiu J, Hindmarch CC, Yao ST, Tasker JG, Murphy D (2011) Transcriptomic Analysis of the Osmotic and Reproductive Remodeling of the Female Rat Supraoptic Nucleus. Endocrinology 152:3483-3491.
- Yagil C, Sladek CD (1990) Osmotic regulation of vasopressin and oxytocin release is rate sensitive in hypothalamoneurohypophysial explants. Am J Physiol 258:R492- 500.
- Mutsuga N, Shahar T, Verbalis JG, Xiang CC, Brownstein MJ, et al. (2005) Regulation of gene expression in magnocellular neurons in rat supraoptic nucleus during sustained hypoosmolality. Endocrinology 146:1254-1267.
- Zhang B, Glasgow E, Murase T, Verbalis JG, Gainer H (2001) Chronic hypoosmolality induces a selective decrease in magnocellular neurone soma and nuclear size in the rat hypothalamic supraoptic nucleus. J Neuroendocrinol 13:29-36.
- Kusano K, House SB, Gainer H (1999) Effects of osmotic pressure and brain-derived neurotrophic factor on the survival of postnatal hypothalamic oxytocinergic and vasopressinergic neurons in dissociated cell culture. J Neuroendocrinol 11:145-152.
- Zeidel ML (2010) Hyponatremia: mechanisms and newer treatments. Endocr Pract 16:882-887.
- Verbalis JG (2010) Brain volume regulation in response to changes in osmolality. Neuroscience 168:862-870.
- Castellano G, Solis-Herruzo JA, Gonzalez A, Morillas JD, Moreno D, et al. (1994) Plasma arginine vasopressin response to oral, gastric, and intravenous water load in patients with cirrhosis. Gastroenterology 106:678-685.
- Komlosi P, Fintha A, Bell PD (2004) Current mechanisms of macula densa cell signalling. Acta Physiol Scand 181:463-469.
- Schnermann J (1998) Juxtaglomerular cell complex in the regulation of renal salt excretion. Am J Physiol 274:R263-279.
- Bai GY, Yuan K, Park WH, Kim SZ, Kim SH (2008) Attenuation of hypoosmotic stress-induced ANP secretion via I(Cl,swell) in renal hypertensive rat atria. Peptides 29:1566-1574.
- Akamatsu N, Inenaga K, Yamashita H (1993) Inhibitory effects of natriuretic peptides on vasopressin neurons mediated through cGMP and cGMP-dependent protein kinase in vitro. J Neuroendocrinol 5:517-522.
- Yamashita H, Inenaga K, Okuya S, Hattori Y, Yamamoto S (1989) Effect of brain-gut peptides upon neurons in centrally regulating sites for drinking. Arch Histol Cytol 52 Suppl:121-127.
- Antunes-Rodrigues J, de Castro M, Elias LL, Valenca MM, McCann SM (2004) Neuroendocrine control of body fluid metabolism. Physiol Rev 84:169-208.
- Velasco Cano MV, Runkle de la Vega I (2010) [Current considerations in syndrome of inappropriate secretion of antidiuretic hormone/syndrome of inappropriate antidiuresis]. Endocrinol Nutr 57 Suppl 2:22-29.
- Sudbury JR, Ciura S, Sharif-Naeini R, Bourque CW (2010) Osmotic and thermal control of magnocellular neurosecretory neurons- -role of an N-terminal variant of trpv1. Eur J Neurosci 32:2022- 2030.
- Grindstaff RR, Grindstaff RJ, Cunningham JT (2000) Effects of right atrial distension on the activity of magnocellular neurons in the supraoptic nucleus. Am J Physiol Regul Integr Comp Physiol 278:R1605-1615.
- Cunningham JT, Bruno SB, Grindstaff RR, Grindstaff RJ, Higgs KH, et al (2002) Cardiovascular regulation of supraoptic vasopressin neurons. Prog Brain Res 139:257-273.
- Antunes-Rodrigues J, Favaretto AL, Gutkowska J, McCann SM (1997) The neuroendocrine control of atrial natriuretic peptide release. Mol Psychiatry 2:359-367.
- Pyner S, Deering J, Coote JH (2002) Right atrial stretch induces renal nerve inhibition and c-fos expression in parvocellular neurones of the paraventricular nucleus in rats. Exp Physiol 87:25-32.
- Dampney RA, Polson JW, Potts PD, Hirooka Y, Horiuchi J (2003) Functional organization of brain pathways subserving the baroreceptor reflex: studies in conscious animals using immediate early gene expression. Cell Mol Neurobiol 23:597-616. 62.
- Takakura AC, Moreira TS, Borella TL, Paulin RF, Colombari DS, et al. (2011) Central mechanisms involved in pilocarpine-induced pressor response. Auton Neurosci [Epub ahead of print, PMID: 21689994].
- Bourque CW, Voisin DL, Chakfe Y (2002) Stretch-inactivated cation channels: cellular targets for modulation of osmosensitivity in supraoptic neurons. Prog Brain Res 139:85-94.
- Day TA, Sibbald JR (1989) A1 cell group mediates solitary nucleus excitation of supraoptic vasopressin cells. Am J Physiol 257:R1020-1026.
- Renaud LP, Jhamandas JH, Buijs R, Raby W, Randle JC (1988) Cardiovascular input to hypothalamic neurosecretory neurons. Brain Res Bull 20:771-777.
- Kang YM, Wang Y, Yang LM, Elks C, Cardinale J, et al. (2010) TNFalpha in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med 222:251-263.
- Knecht K, Michalak A, Rose C, Rothstein JD, Butterworth RF (1997) Decreased glutamate transporter (GLT-1) expression in frontal cortex of rats with acute liver failure. Neurosci Lett 229:201-203.
- Norenberg MD, Huo Z, Neary JT, Roig-Cantesano A (1997) The glial glutamate transporter in hyperammonemia and hepatic encephalopathy: relation to energy metabolism and glutamatergic neurotransmission. Glia 21:124-133.
- Coote JH (2005) A role for the paraventricular nucleus of the hypothalamus in the autonomic control of heart and kidney. Exp Physiol 90:169-173.
- Rossi NF, Maliszewska-Scislo M (2008) Role of paraventricular nucleus vasopressin V1A receptors in the response to endothelin 1 activation of the subfornical organ in the rat. J Physiol Pharmacol 59 Suppl 8:47-59.
- Sunn N, Egli M, Burazin TC, Burns P, Colvill L, et al. (2002) Circulating relaxin acts on subfornical organ neurons to stimulate water drinking in the rat. Proc Natl Acad Sci U S A 99:1701-1706.
- Mogulkoc R, Baltaci AK (2010) Effect of melatonin supplementation on plasma vasopressin response to different conditions in rats with hyperthyroidism induced by L-thyroxine. Regul Pept 161:38-42.
- Velmurugan S, Brunton PJ, Leng G, Russell JA (2010) Circulating Secretin Activates Supraoptic Nucleus Oxytocin and Vasopressin Neurons via Noradrenergic Pathways in the Rat. Endocrinology 151:2681-2688.
- Swart RM, Hoorn EJ, Betjes MG, Zietse R (2011) Hyponatremia and inflammation: the emerging role of interleukin-6 in osmoregulation. Nephron Physiol 118:p45-51.
- Scott V, Brown CH (2011) Kisspeptin Activation of Supraoptic Nucleus Neurons in Vivo. Endocrinology [Epub ahead of print, PMID: 21810945].
- Yosten GL, Pate AT, Samson WK (2011) Neuronostatin acts in brain to biphasically increase mean arterial pressure through sympatho-activation followed by vasopressin secretion: the role of melanocortin receptors. Am J Physiol Regul Integr Comp Physiol 300:R1194-1199.
- Nedvetsky PI, Tamma G, Beulshausen S, Valenti G, Rosenthal W, et al. (2009) Regulation of aquaporin-2 trafficking. Handb Exp Pharmacol 190:133-157.
- Wakamatsu S, Nonoguchi H, Ikebe M, Machida K, Izumi Y, et al. (2009) Vasopressin and hyperosmolality regulate NKCC1 expression in rat OMCD. Hypertens Res 32:481-487.
- Aoyagi T, Koshimizu TA, Tanoue A (2009) Vasopressin regulation of blood pressure and volume: findings from V1a receptordeficient mice. Kidney Int 76:1035-1039.
- Lam TI, Wise PM, OÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¾ÃÂâDonnell ME (2009) Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol 297:C278-289.
- Wallace BK, Foroutan S, OÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¾ÃÂâDonnell ME (2011) Ischemia-induced stimulation of Na-K-Cl cotransport in cerebral microvascular endothelial cells involves AMP kinase. Am J Physiol Cell Physiol 301:C316-326.
- Chen Y, Zhao Z, Hertz L (2000) Vasopressin increases [Ca(2+)](i) in differentiated astrocytes by activation of V1b/V3 receptors but has no effect in mature cortical neurons. J Neurosci Res 60:761- 766.
- Kozniewska E, Romaniuk K (2008) Vasopressin in vascular regulation and water homeostasis in the brain. J Physiol Pharmacol 59 Suppl 8:109-116.
- Meijer E, Boertien WE, Zietse R, Gansevoort RT (2011) Potential deleterious effects of vasopressin IN chronic kidney disease and particularly autosomal dominant polycystic kidney disease. Kidney Blood Press Res 34:235-244.
- Koehler EM, McLemore GL, Tang W, Summy-Long JY (1993) Osmoregulation of the magnocellular system during pregnancy and lactation. Am J Physiol 264:R555-560.
- Neumann I, Russell JA, Landgraf R (1993) Oxytocin and vasopressin release within the supraoptic and paraventricular nuclei of pregnant, parturient and lactating rats: a microdialysis study. Neuroscience 53:65-75.
- Lafarga M, Berciano MT, Del Olmo E, Andres MA, Pazos A (1992) Osmotic stimulation induces changes in the expression of beta-adrenergic receptors and nuclear volume of astrocytes in supraoptic nucleus of the rat. Brain Res 588:311-316.
- Teruyama R, Sakuraba M, Kurotaki H, Armstrong WE (2011) Transient Receptor Potential Channel M4 and M5 in Magnocellular Cells in Rat Supraoptic and Paraventricular Nuclei. J Neuroendocrinol [Epub ahead of print, PMID: 21848647, doi: 10.1111/j.1365-2826.2011.02211.x].
- Li YF, Wang W, Mayhan WG, Patel KP (2006) Angiotensinmediated increase in renal sympathetic nerve discharge within the PVN: role of nitric oxide. Am J Physiol Regul Integr Comp Physiol 290:R1035-1043.
- Powers-Martin K, Phillips JK, Biancardi VC, Stern JE (2008) Heterogeneous distribution of basal cyclic guanosine monophosphate within distinct neuronal populations in the hypothalamic paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 295:R1341-1350.
- Israel JM, Poulain DA, Oliet SH (2010) Glutamatergic inputs contribute to phasic activity in vasopressin neurons. J Neurosci 30:1221-1232.
- Bundzikova J, Pirnik Z, Zelena D, Mikkelsen JD, Kiss A (2008) Response of substances co-expressed in hypothalamic magnocellular neurons to osmotic challenges in normal and Brattleboro rats. Cell Mol Neurobiol 28:1033-1047.
- Llorens-Cortes C, Moos F (2008) Opposite potentiality of hypothalamic coexpressed neuropeptides, apelin and vasopressin in maintaining body-fluid homeostasis. Prog Brain Res 170:559-570.
- Palin K, Moreau ML, Orcel H, Duvoid-Guillou A, Rabie A, et al. (2009) Age-impaired fluid homeostasis depends on the balance of IL-6/IGF-I in the rat supraoptic nuclei. Neurobiol Aging 30:1677-1692.
- Yuan H, Gao B, Duan L, Jiang S, Cao R, et al. (2010) Acute hyperosmotic stimulus-induced Fos expression in neurons depends on activation of astrocytes in the supraoptic nucleus of rats. J Neurosci Res 88:1364-1373.
- Badaut J, Nehlig A, Verbavatz J, Stoeckel M, Freund-Mercier MJ, et al. (2000) Hypervascularization in the magnocellular nuclei of the rat hypothalamus: relationship with the distribution of aquaporin-4 and markers of energy metabolism. J Neuroendocrinol 12:960-969.
- Yool AJ (2007) Aquaporins: multiple roles in the central nervous system. Neuroscientist 13:470-485.
- Espallergues J, Solovieva O, Techer V, Bauer K, Alonso G, et al. (2007) Synergistic activation of astrocytes by ATP and norepinephrine in the rat supraoptic nucleus. Neuroscience 148:712-723.
- Ponzio TA, Wang YF, Hatton GI (2006) Activation of adenosine A2A receptors alters postsynaptic currents and depolarizes neurons of the supraoptic nucleus. Am J Physiol Regul Integr Comp Physiol 291:R359-366.
- Wang Y-F, Hatton GI (2006) Mechanisms underlying oxytocininduced excitation of supraoptic neurons: prostaglandin mediation of actin polymerization. J Neurophysiol 95:3933- 3947.
- Israel JM, Schipke CG, Ohlemeyer C, Theodosis DT, Kettenmann H (2003) GABAA receptor-expressing astrocytes in the supraoptic nucleus lack glutamate uptake and receptor currents. Glia 44:102-110.
- Decavel C, Hatton GI (1995) Taurine immunoreactivity in the rat supraoptic nucleus: prominent localization in glial cells. J Comp Neurol 354:13-26.
- Ponzio TA, Ni Y, Montana V, Parpura V, Hatton GI (2006) Vesicular glutamate transporter expression in supraoptic neurones suggests a glutamatergic phenotype. J Neuroendocrinol 18:253-265.
- Armstrong WE, Rubrum A, Teruyama R, Bond CT, Adelman JP (2005) Immunocytochemical localization of smallconductance, calcium-dependent potassium channels in astrocytes of the rat supraoptic nucleus. J Comp Neurol 491:175-185.
- Park JB, Skalska S, Stern JE (2006) Characterization of a novel tonic gamma-aminobutyric acidA receptor-mediated inhibition in magnocellular neurosecretory neurons and its modulation by glia. Endocrinology 147:3746-3760.
- Fleming TM, Scott V, Joe N, Naskar K, Brown CH, et al. (2011) State-Dependent Changes in Astrocyte Regulation of Extrasynaptic Nmda Receptor Signaling in Neurosecretory Neurons. J Physiol [Epub ahead of print, PMID: 21690192].
- Wang Y-F, Hatton GI (2009) Astrocytic plasticity and patterned oxytocin neuronal activity: dynamic interactions. J Neurosci 29:1743-1754.
- Hatton JD, Ellisman MH (1981) The distribution of orthogonal arrays and their relationship to intercellular junctions in neuroglia of the freeze-fractured hypothalamoneurohypophysial system. Cell Tissue Res 215:309-323.
- Duan L, Yuan H, Su CJ, Liu YY, Rao ZR (2004) Ultrastructure of junction areas between neurons and astrocytes in rat supraoptic nuclei. World J Gastroenterol 10:117-121.
- Wang Y-F, Hamilton K (2009) Chronic vs. acute interactions between supraoptic oxytocin neurons and astrocytes during lactation: role of glial fibrillary acidic protein plasticity. ScientificWorldJournal 9:1308-1320.
- Pasantes-Morales H, Maar TE, Moran J (1993) Cell volume regulation in cultured cerebellar granule neurons. J Neurosci Res 34:219-224.
- Nase G, Helm PJ, Enger R, Ottersen OP (2008) Water entry into astrocytes during brain edema formation. Glia 56:895-902.
- Zhang Z, Bourque CW (2003) Osmometry in osmosensory neurons. Nat Neurosci 6:1021-1022.
- Okada Y, Sato K, Numata T (2009) Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol 587:2141-2149.
- Pasantes-Morales H, Lezama RA, Ramos-Mandujano G, Tuz KL (2006) Mechanisms of cell volume regulation in hypoosmolality. Am J Med 119:S4-11.
- Fraser CL, Swanson RA (1994) Female sex hormones inhibit volume regulation in rat brain astrocyte culture. Am J Physiol 267:C909-914.
- Hussy N, Deleuze C, Desarmenien MG, Moos FC (2000) Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol 62:113-134.
- Park JB, Jo JY, Zheng H, Patel KP, Stern JE (2009) Regulation of tonic GABA inhibitory function, presympathetic neuronal activity and sympathetic outflow from the paraventricular nucleus by astroglial GABA transporters. J Physiol 587:4645- 4660.
- Verbalis JG, Gullans SR (1991) Hyponatremia causes large sustained reductions in brain content of multiple organic osmolytes in rats. Brain Res 567:274-282.
- Middeldorp J, Hol EM (2011) GFAP in health and diseaseProg Neurobiol 93:421-443.
- Anderova M, Kubinova S, Mazel T, Chvatal A, Eliasson C, Pekny M, Sykova E (2001) Effect of elevated K(+), hypotonic stress, and cortical spreading depression on astrocyte swelling in GFAP-deficient mice. Glia 35:189-203.
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, et al. (2004) Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci 24:5016-5021.
- Sobel RA, DeArmond SJ, Forno LS, Eng LF (1981) Glial fibrillary acidic protein in hepatic encephalopathy. An immunohistochemical study. J Neuropathol Exp Neurol 40:625-632.
- Kril JJ, Flowers D, Butterworth RF (1997) Distinctive pattern of Bergmann glial pathology in human hepatic encephalopathy. Mol Chem Neuropathol 31:279-287.
- Vargova L, Chvatal A, Anderova M, Kubinova S, Ziak D, et al (2001) Effect of osmotic stress on potassium accumulation around glial cells and extracellular space volume in rat spinal cord slices. J Neurosci Res 65:129-138.
- Zierler S, Frei E, Grissmer S, Kerschbaum HH (2008) Chloride influx provokes lamellipodium formation in microglial cells. Cell Physiol Biochem 21:55-62.
- Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G (2008) Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des 14:3574-3589.
- Arieff AI (2006) Influence of hypoxia and sex on hyponatremic encephalopathy. Am J Med 119:S59-64.
- Gainer H, Yamashita M, Fields RL, House SB, Rusnak M (2002) The magnocellular neuronal phenotype: cell-specific gene expression in the hypothalamo-neurohypophysial system. Prog Brain Res 139:1-14.
- Miyata S, Hatton GI (2002) Activity-related, dynamic neuronglial interactions in the hypothalamo-neurohypophysial system. Microsc Res Tech 56:143-157.
- Kruczek C, Gorg B, Keitel V, Pirev E, Kroncke KD, et al. (2009) Hypoosmotic swelling affects zinc homeostasis in cultured rat astrocytes. Glia 57:79-92.
- Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou MN, Rapisarda C, et al. (2007) Expression and functional characterization of transient receptor potential vanilloidrelated channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience 148:876-892.
- Borgdorff AJ, Somjen GG, Wadman WJ (2000) Two mechanisms that raise free intracellular calcium in rat hippocampal neurons during hypoosmotic and low NaCl treatment. J Neurophysiol 83:81-89.
- Schliess F, Foster N, Gorg B, Reinehr R, Haussinger D (2004) Hypoosmotic swelling increases protein tyrosine nitration in cultured rat astrocytes. Glia 47:21-29.
- Olson JE, Li GZ, Wang L, Lu L (2004) Volume-regulated anion conductance in cultured rat cerebral astrocytes requires calmodulin activity. Glia 46:391-401.
- Gunnarson E, Zelenina M, Axehult G, Song Y, Bondar A, et al. (2008) Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia 56:587-596. 137.
- Illarionova NB, Gunnarson E, Li Y, Brismar H, Bondar A, et al. (2009) Functional and molecular interactions between aquaporins and Na,K-ATPase. Neuroscience 168:915-925.
- Butterworth RF (2010) Altered glial-neuronal crosstalk: cornerstone in the pathogenesis of hepatic encephalopathy. Neurochem Int 57:383-388.
- Haussinger D, Gorg B, Reinehr R, Schliess F (2005) Protein tyrosine nitration in hyperammonemia and hepatic encephalopathy. Metab Brain Dis 20:285-294.
- Gorg B, Bidmon HJ, Keitel V, Foster N, Goerlich R, et al. (2006) Inflammatory cytokines induce protein tyrosine nitration in rat astrocytes. Arch Biochem Biophys 449:104-114.
- Ebner HL, Fiechtner B, Pelster B, Krumschnabel G (2006) Extracellular signal regulated MAP-kinase signalling in osmotically stressed trout hepatocytes. Biochim Biophys Acta 1760:941-950.
- Carbonell WS, Mandell JW (2003) Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J Neurotrauma 20:327-336.
- Bender AS, Neary JT, Blicharska J, Norenberg LO, Norenberg MD (1992) Role of calmodulin and protein kinase C in astrocytic cell volume regulation. J Neurochem 58:1874-1882.
- Liu YP, Yang CS, Chen MC, Sun SH, Tzeng SF (2010) Ca(2+)- dependent reduction of glutamate aspartate transporter GLAST expression in astrocytes by P2X receptor-mediated phosphoinositide 3-kinase signaling. J Neurochem 113:213- 227.
- Norenberg MD, Rama Rao KV, Jayakumar AR (2009) Signaling factors in the mechanism of ammonia neurotoxicity. Metab Brain Dis 24:103-117.
- 146. Oghlakian G, Klapholz M (2009) Vasopressin and vasopressin receptor antagonists in heart failure. Cardiol Rev 17:10-15.
- Takefuji S, Murase T, Sugimura Y, Takagishi Y, Hayasaka S, et al (2007) Role of microglia in the pathogenesis of osmoticinduced demyelination. Exp Neurol 204:88-94.
- Gankam Kengne F, Soupart A, Pochet R, Brion JP, Decaux G (2009) Re-induction of hyponatremia after rapid overcorrection of hyponatremia reduces mortality in rats. Kidney Int 76:614-621.
- Gankam-Kengne F, Soupart A, Pochet R, Brion JP, Decaux G (2010) Minocycline protects against neurologic complications of rapid correction of hyponatremia. J Am Soc Nephrol 21:2099-2108.
- Jiang J, Sun CW, Alonso-Galicia M, Roman RJ: (1998) Lovastatin reduces renal vascular reactivity in spontaneously hypertensive rats. Am J Hypertens 11:1222-1231.

