Keywords
Prion protein mutation; N-terminal domain; P39L; Tubulin; Proline; Frontotemporal dementia; PRNP
Introduction
Prion proteins
Function: The expression of the normal prion protein (PrP) is widespread in neurons, neuroendocrine cells, and stromal cells of the lymphoreticular system, but the highest levels are found in the central nervous system, notably associated with the synaptic membrane. The conformational conversion of normal cellular prion protein (PrPC) into a protease-resistant, amyloidogenic conformation, PrP Scrapie (PrPSc) is the defining step in prion infection [1] for which expression of PrPC is both required and rate limiting [2,3].
The prion protein is bound to the outer membrane of the cell surface, in specific cholesterol- and glycosphingolipid-rich lipid sites defined as “rafts” [4] by a glycosylphosphatidylinositol (GPI) anchor. After translocation across the endoplasmic reticulum membrane, the N-terminal signal peptide (the first 22 amino acids of the precursor protein) is cleaved [5]. The function of the physiological PrP (PrPC) is still elusive, although it seems to protect against programmed cell death [6]. PrPC is a copperbinding protein with superoxide dismutase activity that appears to protect against oxidative damage [7] and acts as a cell-surface receptor for signal transduction [8]. Several studies have revealed that the mammalian protein is extremely versatile, whereby PrPC is also involved in cell adhesion, proliferation, and differentiation, and in synaptic plasticity [9]. Most of the functions of the PrPC protein are due to its ability to interact with multiple extra- and intra-cellular signaling partners (ligands), with all these signals being advantageous to the cell [10]. Some of these ligands are laminin, glycosamminoglycans (GAGs), involved in neuronal differentiation and axon growth [11], and neuronal adhesion proteins, such as N-CAM12 that leads to neurite outgrowth [12].
Structure, N-terminal, and C-terminal domains: With regards to the structure of the PrP, the mature protein (residues 23-231) can be divided into structurally independent N-terminal (23-120) and C-terminal domains (residues 121-231) [13]. The N-terminal is a flexible, random coil with a disordered amino acid sequence, whereas the C-terminal region forms a more rigid globular domain [14] containing a bundle of three α-helices and a short, two-stranded, antiparallel β-sheet. This domain is stabilized by a disulfide bridge and includes two variably occupied N-linked glycolsylation sites. These elements are arranged into two halves, β1-α1-β2 and α2-α3, which are packed against each other defining the hydrophobic core [13,15-17]. The protein’s structure is conserved across vertebrate classes during evolution and shows a high degree of amino acid sequence similarity [18]. The N-terminal of the PrP (amino acid residues 23-90) harbors insertions and deletions, whereas in the C-terminal portion (91- 231) mainly point mutations are found. It is of note that a high degree of sequence conservation has been identified in the N-terminal region between amino acid residues 23-90 and the regions located upstream of alpha helices 1 and 3 [18].
Mutations in the gene coding the PrP (PRNP), Inherited Prion Diseases, genotype-phenotype correlation, and phenotypic heterogeneity: Pathogenic mutations in the open reading frame (ORF) of the PrP gene (PRNP) are the only cause of Inherited Prion Diseases (IPD) [1,19]. These fatal neurodegenerative disorders follow a dominant mode of inheritance and are traditionally classified clinically as Creutzfeldt-Jacob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and Fatal Familial Insomnia (FFI) [20,21]. PRNP mutations are represented by point mutations leading to an amino acid substitution or premature stop codon, and insertions/deletions of additional (more than three additional) octapeptide repeats (OPRI/OPRD) in the region between codons 51-91 of the PrP that encodes a 5-mer repeat region consisting of a nonapeptide followed by four identical octapeptides.
In addition to these mutations, that appear fully penetrant, many common single nucleotide polymorphisms (SNPs) have also been observed in the ORF of PRNP [22,23], such as the SNP at codon 129, having a critical role in susceptibility and modifier of prion disease, and alterations in the number of repeats, up to three additional repeats. Some pathogenic PRNP mutations are typically associated with particular clinical categories of prion disease [24-26], conferring diagnosis of IPD and a sub-classification according to a specific mutation. In fact, the Gly114Val, Val180Ile, Thr183Ala, Thr188Lys, Glu196Lys, Glu200Lys Val203Ile, Arg208His, Val2010Ile, Glu211Gln, Met232Arg, and Pro238Ser mutations are identified as causative of CJD, whereasthe Pro102Leu, Pro105Thr, Pro105Ser, Ala117Val, Gly131Val, Tyr145Stop, Gln160Stop, His187Arg, Phe198Ser, Asp202Asn, Glu211Gln, Gln212Pro, Gln217Arg, Tyr226Stop, and Gln227Stop genetic variants are associated with GSS. The Asp178Asn mutation accounts for FFI together with the 129Met genotype, whereas the same mutation associated with the 129Val genotype was found in the GSS. However, the distribution and frequency of these mutations can differ between Europeans and East Asians [27,28]. Other mutations are involved in a spectrum of clinical and pathological phenotypes, variable across and within families who are carriers of the same genetic alteration [29] often with striking phenotypic heterogeneity, which may partially depend on the Met129Val polymorphism. This SNP appears to be responsible fora proportion of the variance observed in the age of onset (20-85 years) [19,25] and, in part, in the phenotypic characteristics [25]. In some cases, the clinical picture is not specific and is confined to psychiatric features [24,30]. Moreover, PRNP gene mutations were found to be associated with clinical pictures resembling other neurodegenerative diseases, such as Frontotemporal dementia [31-36], Cerebral amyloid angiopathy (CAA) [37], familial neuropsychiatric illness [38], familial Alzheimer’s disease [39] and Huntington’s disease [24].
The most prevalent missense mutations causing IPD and a series of SNPs are localized in the C-terminal domain [19]. Conversely, in the N-terminal region between codons 51–91 (the region consisting of the octapeptide repeats), alterations (insertions and deletions, OPRI/OPRD) in the number of repeats are found as polymorphisms and pathogenic mutations, however the presence of any pathogenic point mutations in residues 23-50 are unknown to date. The first known residue associated with prion disease is codon 102 (mutation Pro102Leu) that is located near the proteinase K resistant core of the pathogenic prion protein (PrPSc).
The N-terminal domain
The N-terminal domain: The importance of the N-terminal region has largely been overlooked because it does not appear to be essential for prion replication [40] however, several studies have shown that this domain is involved in fibrillation and in the determination of the physical properties of disease-related forms of PrP [41]. The N-terminal region is flexible and largely disordered. Moreover, the high degree of conservation between species of several segments of this flexible domain, including residues 23-90 is remarkable and probably reflects a strong functional significance [18,42].
The N-terminal domain is a disordered region: disordered proteins and their advantages: The lack of stable tertiary and secondary structure offers a variety of functional advantages to intrinsically disordered proteins/regions (IDPs/IPRs): malleability of interaction with different partners (binding promiscuity), specific but low-affinity binding, increased binding rate, and disorder-to-order transition. These characteristics of unstructured, disordered proteins allow for the fine modulation of post-translational modifications, such as phosphorylation, acetylation, acylation, carboxylation, glycosylation, methylation, hydroxylation, etc. These modifications involve low affinity and high specificity interactions between a protein and a specific ligand. Post-translational modifications associated with IDPs and IPRs are especially important for signaling and regulation of the cell (i.e., transcription, DNA repair, signal transduction, autophagy, etc.) [43]. The ability of the PrPC protein to interact with multiple extra- and intra-cellular signaling partners (ligands) is attributed to the rigorous structural disorder of the N-terminal domain, which lies in its specific and not random, conserved, amino acid sequence [44]. In fact, functional changes and susceptibility to prion diseases with various isoforms of prion protein could be caused by numeric variability and conformational changes discovered in this sequence.
Ligands of the N-terminal domain: Natural ligands play a number of roles in the stabilization of proteins and in the modulation of their structures. In fact, during the course of their biological function, proteins undergo different types of structural rearrangements, including local to large-scale conformational changes. These changes are often triggered by protein interactions with low molecular-weight ligands or with larger macromolecules. The interactions with natural ligands can significantly affect protein structure. The possible structural transformations induced in a protein by a ligand vary widely, ranging from a negligible decrease in the conformational stability to complete protein unfolding [43]. The flexible unstructured N-terminal region provides the PrPC with several advantages. The extended linear protein region may allow interaction with many ligands ranging from small molecules (e.g. Cu2+) to macromolecules (e.g. phospholipids, proteins). However, the disordered proteins and their advantages have yet to be described. Binding domains along the entire extent of the PrPC molecule have been identified for a number of natural ligands [45]. Specific ligands of the N-terminal domain include: 1) copper ions that bind at amino acid residues 59–90, which demonstrate an involvement of this region in copper endocytosis and metabolism [46]. Indeed, it has been observed that prion proteins with insertion mutations in this region have altered N-terminal conformation, increased ligand binding activity, and are more susceptible to oxidative attack [47]. 2) Aβ oligomers with high affinity that possibly mediate neurotoxic effects, being the polybasic stretch at the extreme N-terminus one of the two critical regions for the interaction [48,49]. 3) Tubulin: the PrP regions interacting with tubulin have been mapped to the N-terminus of PrP spanning residues 23-50 and 51-91. PrP octapeptide repeats are critical for this binding activity, given that binding becomes stronger as the number of octapeptide repeats increases, thus suggesting a potential role for PrP in regulating microtubule dynamics in neurons [50]. 4) Acetylcholinesterase (AChE), a key protein in the cholinergic system in neural and nonneural tissues. This heterologous association induces aggregation of monomeric PrP and modifies the structural properties of PrP amyloid fibrils. PrP-AChE interaction requires two subsites in the PrP N-terminal domain (residues 23-99 and 100-120) [51].
Functions of specific N-terminal residues: In the PrP, the N-terminal residue is associated with PrPC internalization [52] for which the initial polybasic region (amino acids 23–28 NH2- KKRPKP) has been shown to be especially important [53]. The N-terminal domain (amino acids 23–90) also acts as a rafttargeting signal, as it is sufficient to confer raft localization when fused to a non-raft transmembrane-anchored protein [54]. The polybasic region (amino acids 23–30) seems crucial for the correct folding of the PrPC and may also regulate the acquisition of strainspecific conformations in disease [41]. The region including amino acids 23-50 has been shown to confer a cellular protective effect resulting in reduced intracellular ROS levels [55,56].
Mechanisms causing conformational change of PrP in mammalians: The (C-terminal domain)
How pathogenic mutations in PRNP cause prion disease has yet to be resolved. Despite important advances in the last decade, how PRNP pathogenic mutations play a role in producing a misfoldedPrP remains an open issue. Nevertheless, in an attempt to study the mechanism involved in this conformational rearrangement of the protein, an interesting hypothesis has been proposed relative to PRNP mutations in the C-terminal domain [57,58]. Recent reports have indicated that variation of the PRNP sequence by pathological mutations is sufficient to generate prions [59]. It has been observed that PRNP genetic variations are mostly clustered in the β2-α2-loop region and in the α2-α3 inter-helical interfaces, which are packed against each other defining the hydrophobic core. Different experimental data have suggested that the conformation of the β2-α2-loop plays a role in prion disease transmission and susceptibility. Several studies have indicated that mammals carrying a flexible β2-α2 loop could be easily infected by prions, whereas prions are poorly transmissible to animals carrying a rigid loop [60]. Importantly, the horse and rabbit have so far displayed resistance to prion infections. Some studies have shown that their PrP structures are characterized by a rigid β2-α2 loop and by a closer contact between the loop and the α3 helix [61,62]. Thus, it seems that prion resistance is determined by the amino acidic composition of the β2-α2-loop and its long-range interactions with the C-terminal end of the α3 helix. Using molecular dynamics (MD) simulations of some PRNP mutations, their mutant structure in aqueous solution has been investigated. In contrast to the wild-type protein, the structures of Gln212Pro and Val210Ile mutants point to the interruption of aromatic and hydrophobic interactions between residues located at the interface of the β2-α2 loop and the C-terminal end of α3 helix. A loss of contact between the β2-α2-loop and the α3 helix in the mutants results in higher exposure of hydrophobic residues to solvent. Similar findings have also been reported for Glu200Lys, Phe198Ser, and Asp178Asn mutations. These findings indicatethat the structural disorder of the β2-α2-loop together with the increased distance between the loop and α3 helix represent key pathological structural features and critical epitopes involved in the conversion to PrPSc. Indeed, it seems that the regions most affected by disease-linked mutations in terms of structure and/or flexibility might be those involved in the pathogenic conversion of PrPC to the scrapie form of the protein, and ultimately, in its interaction with cellular partners [57,58]. In fact, it has been demonstrated that the variation in flexibility of the native state of the PrP protein mainly involves residues 165–175 and residues 185–200, comprising the β2-α2-loop and the α2- α3 structural regions, respectively [57]. This flexibility in variation facilitates the access to alternate conformational states of the protein, remodeling the sites involved in molecular recognition events such as protein-protein and protein-ligand interactions [58].
Role of the proline amino acid in the PrP protein
The proline amino acid: Proline is a cyclic, non essential, non polar, amino acid. Proline motifs are known to impart a degree of structure onto proteins due to the steric constraints of the rigid pyrrolidine ring [63]. Proline acts as a structural disruptor in the middle of regular secondary structure elements such as alpha helices and beta sheets. Multiple prolines and/or hydroxyprolines in a row can create a polyproline helix, the poly(L-proline) II (PPII) helix (Figure 1). This (PPII) helix is the predominant secondary structure in proteins with high conformational flexibility, such as collagen, and the presence of proline in the peptide gives its special features like elasticity and tensile strength. The hydroxylation of proline, by prolyl hydroxylase in a hydroxylation reaction, increases the conformational stability of collagen significantly. Hence, the hydroxylation of proline is a critical biochemical process for maintaining the connective tissue of higher organisms. Proline plays important roles in molecular recognition, particularly in intracellular signaling. The domains rich in proline form "pockets" interacting with ligands and are therefore fundamental for intracellular signal transduction. In fact, proteins that possess proline concentrations that are more abundant than other protein sequences are those that are directly involved in signal transduction.
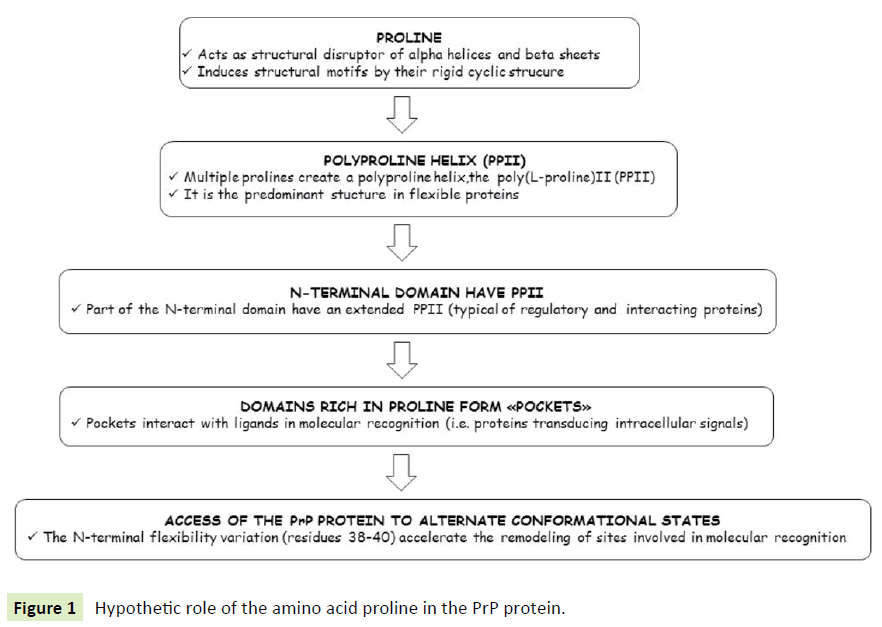
Figure 1 Hypothetic role of the amino acid proline in the PrP protein.
PRNP Pro39Leu mutation and hypothesis for the role of theproline residue in the N-terminal domain: Part of the N-terminal sequence has been predicted to have an extended poly(L-proline) II (PPII) helix structure [64] that has been demonstrated to be involved in regulatory and multiple weak interactions in several proteins [65,66]. This domain has also been implicated in binding to heparan sulfate and other glycosaminoglycans, being important modulators of prion biology [67,68]. Experimental data has demonstrated that the N-terminal residues 37–53 have the potential to form an extended poly(L-proline) II (PPII) helix structure, forming a hydroxylation site at Pro44 [69]. In particular, almost complete conversion of proline to 4-hydroxyproline occurs specifically at residue Pro44 in a murine PrP model. Two sites within the N-terminal segment of PrP match the consensus sequence for enzymatic hydroxylation between residues 27–29 (sequence Lys–Pro–Gly) and 38–40 (sequence Tyr–Pro–Gly). This consensus sequence, and not others, can act as a substrate for prolyl 4-hydroxylation in brain cells of mice infected with prions.
The hypothesized effect of a proline change in the PrP protein: It is possible that the PPII helix N-terminal structure in the PrP protein, as widely demonstrated in other proteins such as collagen, may allow the protein to recognize many different receptors, thereby generating different cellular signals (also signals with opposite activities) depending on substrate structure and/or on binding specificity [70,71]. This extended helix structure, and factors influencing its dynamic flexibility, may be critical for PrP in normal cellular function and signaling.
Moreover, it is also possible that in the PrP protein, such as in the collagen protein, proline residues induce small structural motifs due to the steric constraints imposed by their rigid cyclic structure. Indeed, a mutation of the prolines within the polybasic region, producing a less rigid N-terminus, permitted the peptide to interact more readily with the cell while simultaneously abolishing its specific protective properties and producing potentially deleterious effects [55].
Indeed, as demonstrated in the C-terminal domain [57,58] a variation in flexibility of the native state of the PrP protein involving in this case the N-terminal residues 38-40, may facilitate, by accelerating the access to alternate conformational states of the protein, remodeling of the sites involved in molecular recognition events such as protein-protein, and protein-ligand interactions. Importantly, the detected perturbations can transmit through the protein chain to sites distal to the mutation position [57]. In fact, interactions between proteins and their ligands often result not only in evident local changes in the vicinity of the binding site, but also in global conformational changes. The possible structural transformations induced in a protein by ligand release are very extensive, ranging from a negligible decrease in the conformational stability to complete protein unfolding.
Furthermore, even changes in single amino acids of protein sequences can change its flexibility and consequently the rates at which they aggregate by an order of magnitude of one or more [72,73] thus dramatically accelerating the development of protein depositions and related diseases. In fact, the changes in aggregation rates caused by such mutations have been shown to correlate with changes in simple properties that result from such substitutions, such as charge, secondary structure propensities, and hydrophobicity [73]. Mutations modulate the aggregation propensities of both, well-folded and intrinsically disordered proteins. Numerous neurodegenerative diseases originate from misfolding and neurotoxic aggregation of specific proteins.
Discussion
The speculative hypotheses
The N-terminal PRNP Pro39Leu mutation: For the first time, to our knowledge, a novel missense Pro39Leu mutation in the N-terminal domain of PrP (Figure 2) has been reported in two patients affected by frontotemporal lobar degeneration (FTLD) syndrome [74] and successively in another FTD patient [75] being all the three patients negative for mutations in known causative genes. The absence of this substitution was verified in 200 cognitively healthy controls and indeed, the genetic variation is a mutation and not a common polymorphism. In silico analyses predicted that the mutation is functionally “probably damaging” (PolyPhen-2 score of 1.000), “damaging” (SIFT score of 0.01), and “disease causing” (MutationTaster), respectively [74]. Obviously, functional studies are required to determine whether and how this mutation may exert its pathogenic effects. Herein, we merely attempt to speculate on, based on data reported in the literature, the potential mechanisms that could explain how this mutation may trigger this specific disease phenotype.
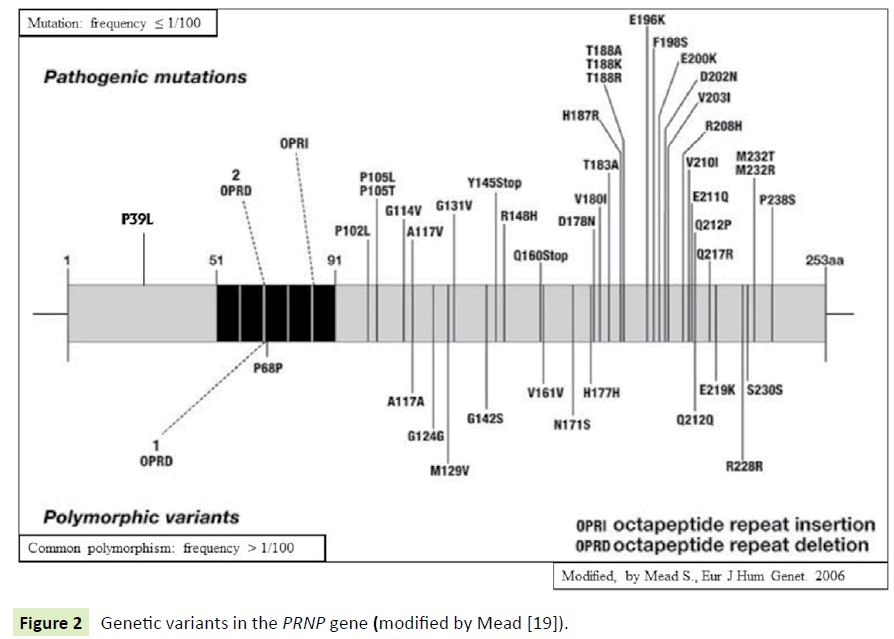
Figure 2 Genetic variants in the PRNP gene (modified by Mead [19]).
There is not a clear relationship between a specific PRNP gene mutation (genotype) and a definite clinical phenotype: Although a correlation was observed between particular PRNP mutations and specific phenotypes (e.g., GSS and Pro102Leu mutation; CJD and Glu200Lys), some of these PRNP mutations have been detected in several other clinical phenotypes that are different from IPD, such as Alzheimer’s disease and Frontotemporal dementia [31-36,39]. This confirms that the search for mutations in the PRNP gene should be considered in these phenotypic manifestations, especially after exclusion of causative mutations in FTD or AD genes.
In contrast, a relationship between a certain PRNP gene mutation (genotype) and a specific neuropathological phenotype has been proven: Atypical clinical phenotypes with mutations in the PRNP gene (such as FTD-like phenotypes) for which neuropathological data were available, have shown the presence of prion disease [31,33,36].
PRNP Pro39Leu mutation-specific pathogenic effects:It has beenhypothesized herein that the Pro39Leu variation, a mutation of a proline within a polybasic region, producing a less rigid N-terminus, might permit peptide-cell interactions more readily and simultaneously abolish its specific protective properties, thus producing potentially deleterious effects [55,69]. Moreover, we might argue that the particular phenotype of our patients, which does not fit the diagnostic criteria of “classical” prion diseases [76] might depend on the location of the mutation in a region not included in the amyloid core of the PrpSc (the disease-related form of PrP), although being biologically active.
Interaction of PrP with tubulin: We speculate that it might be possible that the peculiar FTD-like clinical phenotype presented by these two patients and associated with the PRNP Pro39Leu mutation could depend on the confirmed molecular interaction of PrP with the microtubular cytoskeleton and its major component, tubulin [50] (Figure 3).

Figure 3 Representative pathway of the hypothetical events caused by the Pro39Leu mutation in the PRNP gene.
In fact, the interacting regions within PrP with tubulin have been mapped to the N-terminus of PrP, spanning residues 23- 50 and 51-91. PrP octapeptide repeats are critical for binding activity with tubulin, given that binding activity of PrP with tubulin becomes stronger along as the number of octapeptide repeats increases. These data highlight a potential role of PrP in regulating microtubule dynamics in neurons. Microtubule dynamics are essential in post- mitotic neurons, serving critical roles in axon outgrowth, cell signaling, adhesion, etc. [77]. The presence of many neuronal proteins all serving to control various aspects of microtubule dynamics suggests that a precise regulation of microtubule dynamics is crucial to the development, maintenance, and function of neurons. Microtubule-associated protein tau (MAPT) gene mutations cause a very specific phenotype known as frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), characterized by neuronal cell death and dementia accompanied by abnormal tau fiber pathology. These mutations generally decrease the ability of tau to bind microtubules, increasing its propensity to form abnormal cytotoxic fibers and compromising the ability of the cell to properly regulate microtubule dynamics. Additional evidence supports the idea that destabilization of the microtubule network may be a primary factor in neurotoxicity induced by PrPSc [78-81]. Moreover, an induction of tau hyperphosphorylation by misfolded PrP and a direct interaction between PrP and tau have been demonstrated [82]. Furthermore, concomitant prion pathology and tau-related neurofibrillary degeneration have also been described in the brain tissue of patients carrying PRNP octapeptide repeat insertions. Interestingly, the clinical phenotype of these reported cases differed from typical prion diseases, resembling other forms of dementia such as Alzheimer’s disease or FTD [36,83].
Given that PrP binds directly to tubulin, we presume that it is possible that a mutation in the binding region, such as Pro39Leu, could cause a perturbation in the regulation of microtubule dynamics in mutant neurons, leading to a neurodegenerative FTD-like clinical phenotype.
In contrast, it is also possible that this FTD-like phenotype may depend on the early involvement of topographic pathology in frontal regions compared to other brain regions. Although we are certain that the PRNP Pro39Leu is not a common polymorphism (verified in 200 cognitively healthy controls) we cannot exclude that this genetic variant might be a very rare polymorphism. In this case, the genetic variant may act as a risk factor predisposing to neurodegeneration.
Conclusion
In this paper, given the total absence of neuropathological and functional data so far, we have taken the opportunity to investigate the hypothetical pathogenic mechanism of the novel PRNP Pro39Leu mutation, by reviewing, based on published data,the pathogenic mechanisms of the PRNP mutations and comparing the biochemical properties of the PrP C- and N-terminal domains. Successively, giving greater emphasis to the N-terminal domain, to date largely overlooked, we speculated that the pathogenicity of the PRNP Pro39Leu mutation may depend on its location in the N-terminal domain. We feel that our hypotheses, based only on published data and albeit speculative, may awakened a surge of interest in research specifically targeting the N-terminal domain and a possible interaction with tubulin in causing peculiar phenotypes, such as the FTD-like phenotype. Obviously, we hope to read soon published functional and neuropathological studies determining whether and how variations in this domain might trigger the extreme phenotypic variability associated with the PrP protein and that could confirm, or not, our speculative hypothesis.
Acknowledgments
We thank all subjects and families participating to the study and the Associazione per la Ricerca Neurogenetica-ONLUS Lamezia Terme for invaluable help in assisting persons and families.
19924
References
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93: 337-348.
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, et al. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339-1347.
- Weissmann C, Raeber AJ, Montrasio F, Hegyi I, Frigg R, et al. (2001) Prions and the lymphoreticular system. Philos Trans R Soc Lond B Biol Sci 356: 177-184.
- Taylor DR, Hooper NM (2006) The prion protein and lipid rafts. Mol Membr Biol 23: 89-99.
- Van Rheede T, Smolenaars MM, Madsen O, De Jong WW (2003) Molecular evolution of the mammalian prion protein. Mol Biol Evol 20: 111-121.
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, et al. (1999) Prions prevent neuronal cell-line death. Nature 400: 225-226.
- Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, et al. (1999) Normal prion protein has an activity like that of superoxide dismutase. Biochem J 344: 1-5.
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, et al. (2000) Signal transduction through prion protein. Science 289: 1925-1928.
- Sorgato MC, Peggion C, Bertoli A (2009) Is, indeed, the prion protein a Harlequin servant of "many" masters? Prion 3: 202-205.
- Aguzzi A, Sigurdson C, Heikenwaelder M (2008) Molecular mechanisms of prion pathogenesis Annu Rev Pathol 3: 11-40.
- Hundt C, Peyrin JM, Haik S, Gauczynski S, Leucht C, et al. (2001) Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J 20: 5876-5886.
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 169: 341-354.
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, et al. (1996) NMR structure of the mouse prion protein domain PrP(121-231). Nature 382: 180-182.
- Wüthrich K, Riek R (2001) Three-dimensional structures of prion proteins. Adv Protein Chem 57: 55-82.
- Zahn R, Liu A, Luhrs T, Riek R, Von Schroetter C, et al. (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97: 145-150.
- Calzolai L, Lysek DA, Guntert P, Von Schroetter C, Riek R, et al. (2000) NMR structures of three single-residue variants of the human prion protein. Proc Natl Acad Sci USA 97: 8340-8345.
- Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K (2005) Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA 102: 646-650.
- Wopfner F, Weidenhöfer G, Schneider R, Von Brunn A, Gilch S, et al. (1999) Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 289: 1163-1178.
- Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P (2011) Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121 :21-37.
- Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P (2003) Hereditary prion protein amyloidoses. Clin Lab Med 23: 65-85.
- Mastrianni JA (2010) The genetics of prion diseases. Genet Med 12: 187-195.
- Lloyd S, Mead S, Collinge J (2011) Genetics of prion disease. Top Curr Chem 305: 1-22.
- Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, et al. (1992) Inherited prion disease with 144 base pair gene insertion: Clinical and pathological features. Brain 115: 687-710.
- Wadsworth JD, Hill AF, Beck JA, Collinge J (2003) Molecular and clinical classification of human prion disease. Br Med Bull 66: 241-254.
- Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H (2002) Mutations of the prion protein gene phenotypic spectrum. J Neurol 249(11):1567-1582.
- Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, et al. (2005) Genetic prion disease: The EUROCJD experience. Hum Genet 118: 166-174.
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, et al. (2010) Prospective 10-year surveillance of human prion diseases in Japan. Brain 133: 3043-3057.
- Parchi P, Strammiello R, Giese A, Kretzschmar H (2011) Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol 121: 91-112.
- Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, et al. (1999) Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain 122: 2375-2386.
- Nitrini R, Teixeira da Silva LS, Rosemberg S, Caramelli P, Carrilho PE, et al. (2001) Prion disease resembling frontotemporal dementia and parkinsonism linked to chromosome 17. Arq Neuropsiquiatr 59: 161-164.
- Hall DA, Leehey MA, Filley CM, Steinbart E, Montine T, et al. (2005) PRNP H187R mutation associated with neuropsychiatric disorders in childhood and dementia. Neurology 64: 1304-1306.
- Woulfe J, Kertesz A, Frohn I, Bauer S, George-Hyslop PS, et al. (2005) Gerstmann-Sträussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol 110: 317-319.
- Clerici F, Elia A, Girotti F, Contri P, Mariani C, et al. (2008) Atypical presentation of Creutzfeldt-Jakob disease: the first Italian case associated with E196K mutation in the PRNP gene. J Neurol Sci 275: 145-714.
- Giovagnoli AR, Di Fede G, Aresi A, Reati F, Rossi G, et al. (2008) Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol Sci 29: 405-410.
- Kumar N, Boeve BF, Boot BP, Orr CF, Duffy J, et al. (2011) Clinical characterization of a kindred with a novel 12-octapeptide repeat insertion in the prion protein gene. Arch Neurol 68: 1165-1170.
- Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, et al. (1996) Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA 93: 744-748.
- Samaia HB, Mari JJ, Vallada HP, Moura RP, Simpson AJ, et al. (1997) A prion-linked psychiatric disorder. Nature 390: 241.
- Finckh U, Muller TT, Mann U, Eggers C, Marksteiner J, et al. (2000) High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 66: 110–117.
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, et al. (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255-1264.
- Ostapchenko VG, Makarava N, Savtchenko R, Baskakov IV (2008) The polybasic N-terminal region of the prion protein controls the physical properties of both the cellular and fibrillar forms of PrP. J Mol Biol 383: 1210-1224.
- Kim Y, Lee J, Lee C (2008) In silico comparative analysis of DNA and amino acid sequences for prion protein gene. Transbound Emerg Dis 55: 105-114.
- Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, et al. (2007) Functional anthology of intrinsic disorder. 3 Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res 6: 1917-1932.
- Liu Z, Huang Y (2014) Advantages of proteins being disordered. Protein Sci 23: 539-550.
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, et al. (2008) Physiology of the prion protein. Physiol Rev 88: 673-728.
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, et al. (1997) The cellular prion protein binds copper in vivo. Nature 390: 684-687.
- Yin S, Yu S, Li C, Wong P, Chang B, et al. (2006) Prion proteins with insertion mutations have altered N-terminal conformation and increased ligand binding activity and are more susceptible to oxidative attack. J Biol Chem 281: 10698-10705.
- Chen S, Yadav SP, Surewicz WK (2010) Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 285: 26377-26383.
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457: 1128-1132.
- Dong CF, Shi S, Wang XF, An R, Li P, et al. (2008) The N-terminus of PrP is responsible for interacting with tubulin and fCJD related PrP mutants possess stronger inhibitive effect on microtubule assembly in vitro. Arch Biochem Biophys 470: 83-92.
- Torrent J, Vilchez AA, Munoz TD, Trovaslet M, Nachon F, et al. (2015) Interaction of prion protein with acetylcholinesterase: potential pathobiological implications in prion diseases. Acta Neuropathol Commun 3: 18.
- Nunziante M, Gilch S, Schatzl HM (2003) Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem 278: 3726-3734.
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, et al. (2003) The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 22: 3591-3601.
- Walmsley AR, Zeng F, Hooper NM (2003) The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J Biol Chem 278: 37241-37248.
- Haigh CL, Drew SC, Boland MP, Masters CL, Barnham KJ, et al. (2009) Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. J Cell Sci 122: 1518-1528.
- Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA (2011) The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One 6: e25675.
- Meli M, Gasset M, Colombo G (2011) Dynamic diagnosis of familial prion diseases supports the β2-α2 loop as a universal interference target. PLoS One 6: e19093.
- Legname G (2012) Early structural features in mammalian prion conformation conversion. Prion 6: 37-39.
- Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, et al. (2009) Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63: 438-450.
- Sigurdson CJ, Nilsson KP, Hornemann S, Manco G, Fernández-Borges N, et al. (2010) A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest 120: 2590-2599.
- Perez DR, Damberger FF, Wüthrich K (2010) Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol 400: 121-128.
- Wen Y, Li J, Yao W, Xiong M, Hong J, et al. (2010) Unique structural characteristics of the rabbit prion protein. J Biol Chem 285: 31682-31693.
- Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpe S (1995) Proline motifs in peptides and their biological processing. FASEB J 9: 736-744.
- Smith CJ, Drake AF, Banfield BA, Bloomberg GB, Palmer MS, et al. (1997) Conformational properties of the prion octa-repeat and hydrophobic sequences. FEBS Lett 1405: 378-384.
- Siligardi G, Drake AF (1995) The importance of extended conformations and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers 37: 281-292.
- Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 293: 321-331.
- Gabizon R, Meiner Z, Halimi M, Ben-Sasson SA (1993) Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J Cell Physiol 157: 319-325.
- Caughey B, Brown K, Raymond GJ, Katzenstein GE, Thresher W (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red. J Virol 68: 2135-2141.
- Gill AC, Ritchie MA, Hunt LG, Steane SE, Davies KG, et al. (2000) Post-translational hydroxylation at the N-terminus of the prion protein reveals presence of PPII structure in vivo. EMBO J 19: 5324-5331.
- Tompa P, Szász C, Buday L (2005) Structural disorder throws new light on moonlighting. Trends Biochem Sci 30: 484-489.
- Béland M, Roucou X (2012) The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem 120: 853-868.
- Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24: 329-332.
- Dobson CM (2004) Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 15: 3-16.
- Bernardi L, Cupidi C, Frangipane F, Anfossi M, Gallo M, et al. (2014) Novel N-terminal domain mutation in prion protein detected in 2 patients diagnosed with frontotemporal lobar degeneration syndrome. Neurobiol Aging 35: 2657.e7-11.
- Oldoni E, Fumagalli GG, Serpente M, Fenoglio C, Scarioni M, et al. (2016) PRNP P39L variant is a rare cause of frontotemporal dementia in italian population. J Alzheimers Dis 50: 353-357.
- Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, et al. (2009) Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132: 2659-2668.
- Zhou FQ, Snider WD (2005) Cell biology. GSK-3beta and microtubule assembly in axons. Science 308: 211-244.
- Guo Y, Gong HS, Zhang J, Xie WL, Tian C, et al. (2012) Remarkable reduction of MAP2 in the brains of scrapie-infected rodents and human prion disease possibly correlated with the increase of calpain. PLoS One 7: e30163.
- Li XL, Wang GR, Jing YY, Pan MM, Dong CF, et al. (2011) Cytosolic PrP induces apoptosis of cell by disrupting microtubule assembly. J Mol Neurosci 43: 316-325.
- Wang GR, Shi S, Gao C, Zhang BY, Tian C, et al. (2010) Changes of tau profiles in brains of the hamsters infected with scrapie strains 263 K or 139 A possibly associated with the alteration of phosphate kinases. BMC Infect Dis 10: 86.
- Zhou RM, Jing YY, Guo Y, Gao C, Zhang BY, et al. (2011) Molecular interaction of TPPP with PrP antagonized the CytoPrP-induced disruption of microtubule structures and cytotoxicity. PLoS One 6: e23079.
- Osiecka KM, Nieznanska H, Skowronek KJ, Jozwiak J, Nieznanski K (2011) Tau inhibits tubulin oligomerization induced by prion protein. Biochim Biophys Acta 1813: 1845-1853.
- Kaski DN, Pennington C, Beck J, Poulter M, Uphill J, et al. (2011) Inherited prion disease with 4-octapeptide repeat insertion: disease requires the interaction of multiple genetic risk factors. Brain 134: 1829-1838.

