Keyword
Porcine reproductive and respiratory syndrome virus, Anti-PRRSV peptide
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) is a main cause of porcine reproductive and respiratory syndrome (PRRS), which is a wide-spread infectious disease of farm pigs [1]. Currently, the prevention and treatment of PRRS are becoming a more and more challenging research topic in veterinary viral immunology. However, the specific mechanism of immune response against PRRS virus (PRRSV) still remains unknown [2]. At early stage of infections, pigs develop a rapid but strong humoral response. These initial immune reactions, however, do not bring about protection and may even cause harm through mediating an antibody-dependent enhancement of disease. Neutralizing these adverse antibodies is a slow process and the cell-mediated immune responses are always suppressed by the virus. So an effective vaccine against PPRSV is in great need, but the fact is that the development of vaccine often stuck into dilemma because of high diversity of the virus genome. Increasing evidences show that the immunoreactions induced by one strain may be less or little effective to another different strain, even within the same genotype. Besides, the effect of commercial vaccines against PRRSV is quite limited [2]. And there is a potential risk that the modified-live PRRSV vaccine might revert to virulent virus under farm conditions, which poses great safety concern. Indeed, it has been demonstrated that a conversion from a commercial modified-live PRRSV vaccine to pathogenic phenotype in vaccinated pigs did happened [3-6]. Therefore, it is urgent to explore new antiviral method to control the PRRS. So searching for the virus inhibitors as drugs might be preferred.
PRRSV is divided into two major genotypes: the European (EU; type 1) genotype and the North American (NA; type 2) genotype [7]. The latter is the main type isolated in China. PRRSV is a single-stranded positive-sense RNA virus with 15KB length of genome. The genome is compose of a cap structure at 5′-end and a poly(A) tail at 3′-end, containing nine open reading frames (ORF1a, ORF1b, ORF2a, ORF2b, ORF3–ORF7). ORF1 and ORF2 encode virus replication-related proteins, and account for 80% of the virus genome [8,9]. ORF1a and ORF1b encode polyproteins, which are proteolytic processed into 12 non-structural proteins (NSPs). Among these, proteins from ORF1b perform as the RNAdependent RNA polymerase (NSP9), helicase (NSP10) and the conserved C-terminal domain (CTD) (NSP11) [10], which are important factors in the process of PRRSV infection [11]. Given the facts above, ORF1b can be treated as a potential target for antiviral drug screening [12-16].
Phage technology is an effective method for developing new drugs, and it has been applied in drug development trials increasingly [12,17,18]. Several successful cases of PRRSV inhibitors through phage screening technology were reported. Peptides obtained from phage screened of purified PRRSV showed low inhibition effect against PRRSV [19]. Except for antiviral peptides screen, phage was also used for pathogen detection [20]. In previous study, we obtained several peptides from phage display screen against PPRSV polymerase [21], instead of the whole virus. One of these peptides, P3 showed higher antiviral activity in vitro and lower cytotoxicity in vivo. Properties assay indicated that P3’s strong solubility and positive charge make itself exhibit cationic anti-bacterial peptide characters. These evidences indicate that P3 has the potential possibility to become an antiviral peptide drug.
Methods
Cells and virus
Monkey Kidney cells (MARC-145 ATCC No. CRL-12231) were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS, Gibco). PRRSV polymerase and helicase genes were cloned from PRRSV SY0608 strain. pAPRRSV (Genbank No. GO330474) and pSHE (Genbank No. GO461593) strains were kindly gift from Dr. Wei.
Peptides synthesize
All peptides in this study were chemically synthesized by TASH Biotechnology Co. Ltd with purity of more than 95%, and were dissolved in DMSO.
Real-time polymerase chain reaction (PCR) and tissue culture infective dose (TCID50) assay
MARC-145 cells were inoculated with viruses at 0.01 MOI for 1.5 h at 37°C, and treated with or without peptides at different concentration. The effects on PRRSV of selected peptides treatment were determined by both real-time PCR and TCID50 methods. Simply, total RNA was prepared using TRizol reagent (Invitrogen) from cells treated with or without peptides or drugs. Reverse Transcription was performed following manufacturer protocols (Invitrogen). Briefly, 2 μl total RNA, 1 μl 10 mM dNTP Mix, 2 pmol gene-specific reverse primer and 8.8 μl distilled water were added to a nuclease-free micro-centrifuge tube. Mixtures were heated to 65°C for 5 min and quickly chilled on ice for 5 min. Then 4 μl 5X First-Strand Buffer, 2 μl 0.1 M DTT, 1 μl Ribonuclease Inhibitor and 1 μl M-MLV were added. Tubes were incubated at 42°C for 50 min and 75°C for 15 min. To remove RNA complemented with cDNA, 1 μl E. Coli RNase H was added and incubate at 37°C for 20 min. Real-time PCR was performed using Taqman probe quantitative real-time PCR following manufacturer protocols (TaKaRa). The primers used for PCR are shown in Table 1. Results are expressed as virus RNA copies, which were normalized to β-actin copy numbers as the endogenous control [22].
| Real time PCR Primers |
Sequences (5’-3’) |
| PRRSV F |
AGTGGGTCGGCACCAGTT |
| PRRSV R |
GCAGACAAATCCAGAGGCTCAT |
| MARC-145 actin F |
CAGCACGATGAAGATCAA |
| MARC-145 actin R |
GGGTGTAACGCAACTAAG |
Table 1: Primers for Real time PCR, primers and probes used in this study.
TCID50 was determined by Reed-Muench method according to the previous report [23]. Briefly, MARC-145 cells were plated into a 96-well plate, 0.01 MOI PRRSV was added to 96- well plate containing a monolayer of MARC-145 cells for 1.5 h. After inoculation, peptide or DMSO was added. 24 h later, supernatant was collected and frozen at −70°C until use. Serial 10-fold dilutions were made of supernatant stocks, and 100 μl samples of each dilution were added to duplicate wells of a 96- well plate containing a confluent monolayer of MARC-145. Seven days were allowed for the appearance of cytopathology. The dilution causing cytopathology in half the cultures (the median tissue culture infective dose called TCID50) was then calculated as described by Reed and Muench method.
Cytotoxicity assay
CC50 was determined using MTT assay. Briefly, MARC-145 cells were incubated in 96-well plates in 200 μL of DMEM. MARC- 145 cells were incubated with or without peptides (from 1-250 μM) for 24h. Incubation was terminated by aspirating the media and MTT solution (5 mg/ml in PBS) was added to each well. Formazane formation was terminated after 4 h by removing the MTT solution. Subsequently DMSO was added to each well to solubilize formazane, and the formazane-containing samples were measured at 590 nm in a microplate reader.
Absorption kinetics experiments
MARC-145 Cells were digested to single cell and resuspended in DMEM supplemented with 10% FBS. Approximately 1 x 106 cells were mixed with rhodamine labeled P3 in a culture dish (final concentration of rhodamine labeled P3 was 250 μM ) and incubated in 37°C for different times. Cells were shaken in 20 min time interval. After incubation, cells were washed in phosphate buffered saline (PBS) and lysed in lysis buffer. Lysed supernatant was separated by centrifugation at 5,000 g for 10 min. 100 μL lysed supernatant was added to wells of 96-well plate.
Thereafter, the plate was transferred to BioTekSynergyTM2 and the fluorescence was measured (excitation wavelength 530/25 nm, emission wavelength 528/20 nm). The results were normalized with cells without treatment. Data was calculated and assayed by GEN 5.
Confocal fluorescence microscopy
MARC-145 cells were treated with or without peptide at different time points. After fixation in 80% ice-cold acetone at -20°C for 10 min, cells were stained with 4',6-diamidino-2-phenylindole (DAPI) for 10 min. After washing, cells were detected and photographed with confocal fluorescence microscopy.
Bimolecular fluorescence complementation (BiFC) assay
A variant of yellow fluorescent protein plasmid, Venus, was obtained by reforming of pEYFP-N1 (Genbank Accession: U55762.1). Four polyclone sites (Bgl II, EcoRI, BamHI and XhoI) and (GGGGS)x3 linker were added in front of EYFP first. Then, EYFP was truncated in N-terminus part (VC 174-239) or C-terminus part (VN 1-173), respectively. As shown in Figure 4a, target gene fragments were fused to truncated EYFP via a (GGGGS) X 3 linker [24,25]. Helicase gene from PRRSV was amplified and fused to the N-terminus of VN and mutant P3 (mP3) (STQRPTLMRTRP) was amplified and fused to the N-terminus of VC, which was treated as negative controls. All genes were cloned to plasmid by Bgl II and XhoI sites. The primers used for BiFC are shown in Table 1. About 60% confluency of MARC-145 cells cultured in 6-well plates were co-transfected with 350 ng of each BiFC plasmid using liposome 2000 Transfection Reagent. After 18h post-transfection, living cells were visualized through an Olympus inverted fluorescence microscope.
Statistical analyses
All assays described here were repeated at least twice, and all the measurements were made in triplicate. Mean values±SD (Standard Deviation) were calculated using Microsoft Excel. Student’s t-test or one-way analysis of variance was used to test for significant differences between means, with P<0.05 being considered statistically significant. Figures were performed using the GraphPadTM Prism 5.0 software.
Results
Compare of antiviral peptides against PRRSV
As described before, proteins encoded by PRRSV ORF1b play vital roles during PRRSV replication. In our previous study, we had used phage screening technology to screen out several effective peptides targeting ORF1b proteins. Among these peptides, SPHIIRNHRLSK (P3) exhibited highest antiviral activity. To further explore P3’s comprehensive antiviral ability, we compared the physical properties of it. P3’s IC50 is 67.79μM, determined by Realtime PCR assay. P3 also showed strong solubility (GRAVY-1.100) and positive charge (PI 12.01) in culture media. Strong solubility indicated that P3 is easy to be absorbed at a high concentration in vivo and in vitro. Positive charge of P3 implied it performed antiviral effect just like cationic anti- microbial peptide (Table 2). These two properties facilitated P3’s inhibition effect on PRRSV in vitro.
| Name |
Amino acid sequence |
MW |
pI |
GRAVY |
IC50 (µM) |
CC50 (µM) |
SI |
| P3 |
SPHIIRNHRLSK |
1457.7 |
12.01 |
–1.100 |
67.79 |
618.73 |
9.12 |
Table 2: Antiviral activity and cytotoxicity of p3 peptide against PRRSV. The molecular weight (MW), pI, and grand average of hydropathicity (GRAVY) were predicted with the ProtParam algorithm. IC50: peptide concentration required to inhibit virus infection by 50%; CC50: peptide concentration required to reduce cell viability by 50%, as determined by the MTT method; SI (selectivity index) = CC50/IC50.
Inhibitory effect of P3 peptide against PRRSV infection in vitro
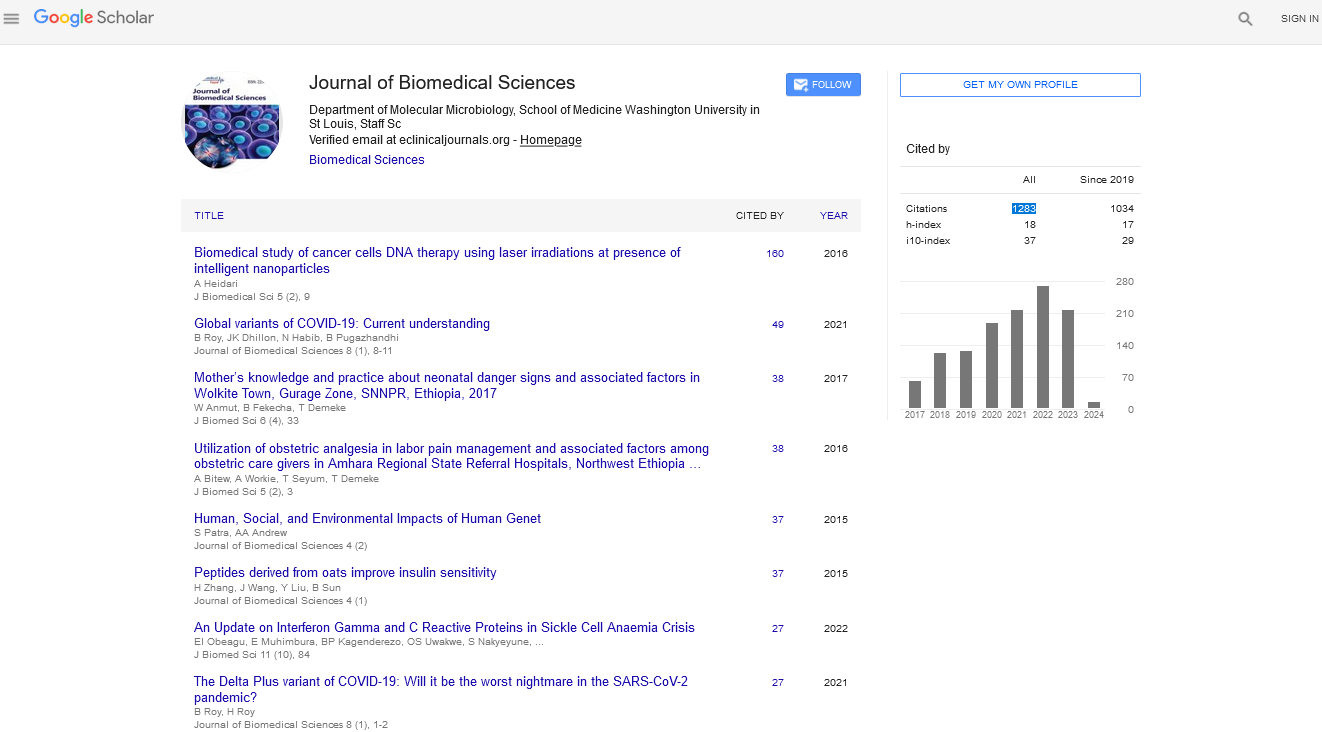
The inhibition ability of P3 was determined by real-time PCR. MARC-145 cells were incubated with viruses at 0.01 MOI for 1.5 h at 37°C, and then treated with P3 peptides at 250 μM for 24 h. The cells treated with DMSO were used as the negative control. P3 showed high antiviral activity in MARC-145 cells, meanwhile DMSO showed no activity against PRRSV (Figure 1a). The results indicated that P3 peptide exerted high antiviral activity in vitro, and the antiviral activity was dependent on the amino acid structure of P3. TCID50 showed strong inhibition of P3 against PRRSV (Figure 1b). The results of Real-time PCR and TCID50 showed that P3 was an effective antiviral peptide against PRRSV in vitro.
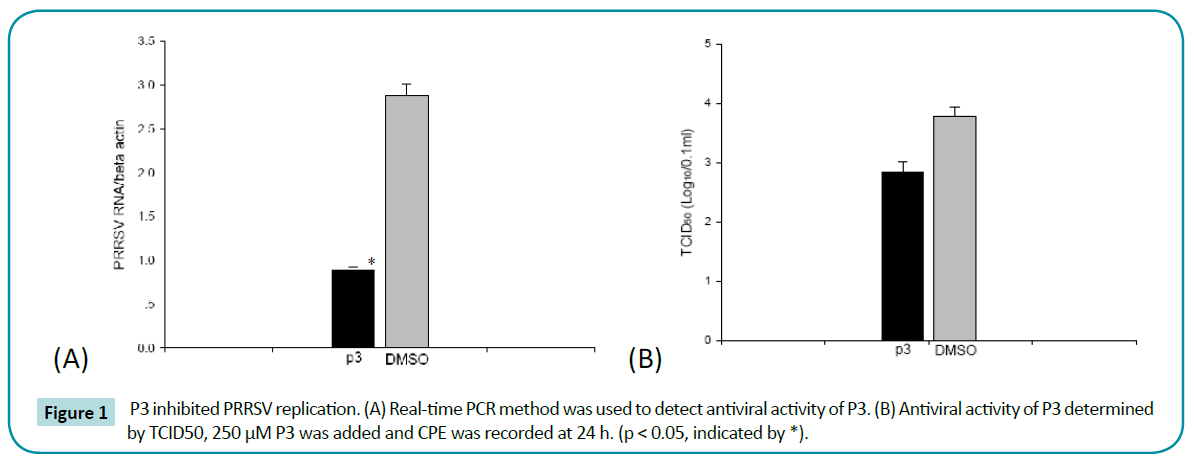
Figure 1: P3 inhibited PRRSV replication. (A) Real-time PCR method was used to detect antiviral activity of P3. (B) Antiviral activity of P3 determined by TCID50, 250 μM P3 was added and CPE was recorded at 24 h. (p < 0.05, indicated by *).
Time- and dose-course test of antiviral activity.
To further investigate the antiviral activity of P3, time-course effect of P3 on inhibition of PRRSV replication was performed. As shown in Figure 2a, during PRRSV replication (12 - 48 h), P3 showed significant inhibition on PRRSV replication compare to control. Furthermore, P3 inhibited PRRSV replication by a dose-dependent manner. The intracellular PRRSV numbers were gradually decreased when peptides concentrations were gradually increased (Figure 2b). These results suggested that P3 exhibited inhibition on PRRSV replication in a time and dosedependent manner.
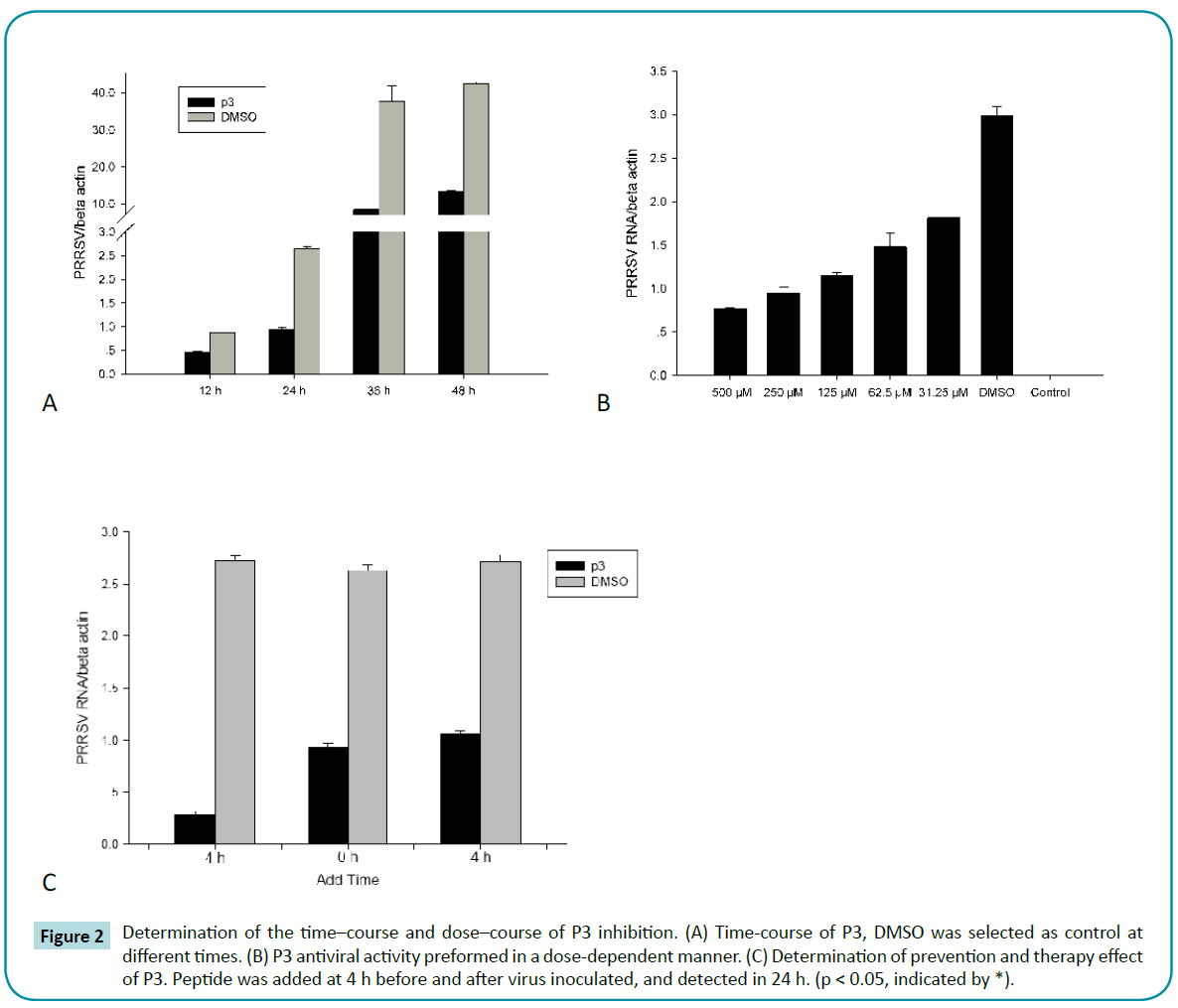
Figure 2: Determination of the time–course and dose–course of P3 inhibition. (A) Time-course of P3, DMSO was selected as control at different times. (B) P3 antiviral activity preformed in a dose-dependent manner. (C) Determination of prevention and therapy effect of P3. Peptide was added at 4 h before and after virus inoculated, and detected in 24 h. (p < 0.05, indicated by *).
P3 exert its prevention and therapy effect through inhibiting PRRSV replication.
To explore the prevention and therapy effect of P3, we treated MARC-145 cells with peptide for 4 h before and after virus infection, respectively. The Real-time PCR results showed that pre-incubated with P3 for 4 h, the intracellular PRRSV numbers was violently decreased after 24 h infection, compared to control which was pre-incubated with DMSO (Figure 2c). Although PRRSV RNA numbers in infected cells treated with P3 were lower than that of DMSO control, it was still significantly higher than that of pre-incubated with P3 for 4 h. These results suggested that P3 pre-incubate before virus infection could strongly inhibit PRRSV replication in MARC-145 cells.
Absorption kinetics of P3
As a virus polymerase inhibitor, it is important to be absorbed by cells to execute its antiviral function. To monitor the process of uptake, the labeled P3 was incubated with MARC-145 cells for 1 hour. Cells were washed and then incubated for another 120 minutes before fixation for confocal fluorescence microscopy (Figure 3a). The results showed that the P3’s uptake efficiency was very high. Basically all the cells showed fluorescence throughout cytoplasm and nucleus. Meanwhile, we measured intracellular concentration of labeled P3 after different incubation. Surprisingly, P3 could penetrate into cells in a very short time after exposure to cells at 37°C (Figure 3b). In the same way, we examined the stability of P3 in MARC-145 cells. After 24 hours exposure, P3 still maintained high level of concentration in vivo (Figure 3c).
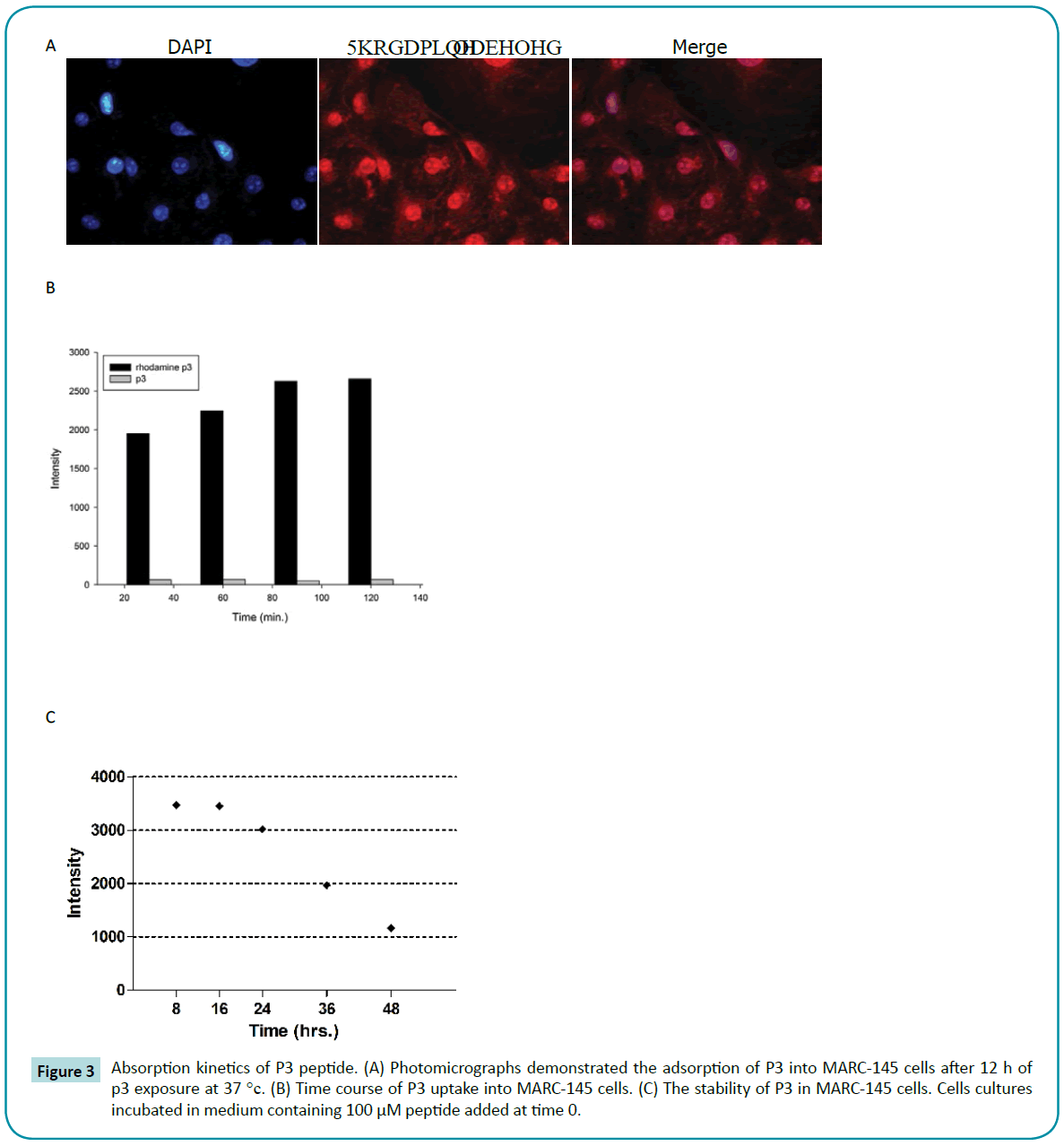
Figure 3: Absorption kinetics of P3 peptide. (A) Photomicrographs demonstrated the adsorption of P3 into MARC-145 cells after 12 h of p3 exposure at 37 °c. (B) Time course of P3 uptake into MARC-145 cells. (C) The stability of P3 in MARC-145 cells. Cells cultures incubated in medium containing 100 μM peptide added at time 0.
P3 interacted with PRRSV polymerase in cells directly
To further demonstrate that P3 could directly bind with polymerase intracellular, the biomolecular fluorescence complementation (BiFC) had been conducted. The principle of BiFC assay is based on structure complementation between two non-fluorescent Nand C-terminal fragments of an intact fluorescent protein. If the proteins of interest do interact, the non-fluorescent fragments can be brought into close proximity and the fluorescence of intact fluorescent protein can be visualized using fluorescence microscopy. BiFC assay allows for the visualization of specific protein-protein interactions within the living cells (Figure 4a).
The fluorescent protein between amino acid residues 173 and 174 was split to generate two fragments, VN and VC. As expected, neither VN nor VC expressed alone produced a fluorescent signal when expressed in MARC-145 cells (data not shown). Likewise, co-expression of VN and VC yet also produced no fluorescence in MARC-145 cells (Figure 4b). We then generated full PRRSV polymerase and helicase gene to fuse into the N-terminus of VN with a flexible linker and designated VN-polymerase and VNhelicase, respectively. And we generated P3 and mutant P3 gene (HIRHRKSIPSNL) to fuse into N-terminus of VC with a flexible linker and designated VC-P3 and VC-mP3. In order to determine whether BiFC can efficiently detect the polymerase-P3 interactions in living cells, cells were co-transfected with the different combinations of VN-polymerase, VN-helicase, VC-P3 and VC-mP3, and ultimately were examined for fluorescence. After 18 hours transfection, as expected, VN and VC co-expression produced no fluorescence signal. Meanwhile, the combination of VN-helicase and VCP3 also failed to generate fluorescence, demonstrating that P3 couldn’t bind with PRRSV helicase. In the pairs of VN-polymerase and VC-P3, fluorescence was observed in the cytoplasm (Figure 4b), representing the p9-polymerase strong interaction. When P3 was mutated, no positive BiFC signals had been captured. The VN-helicase/VC-P3 and VN-polymerase/VC-mP3 pairs were employed as negative controls here.
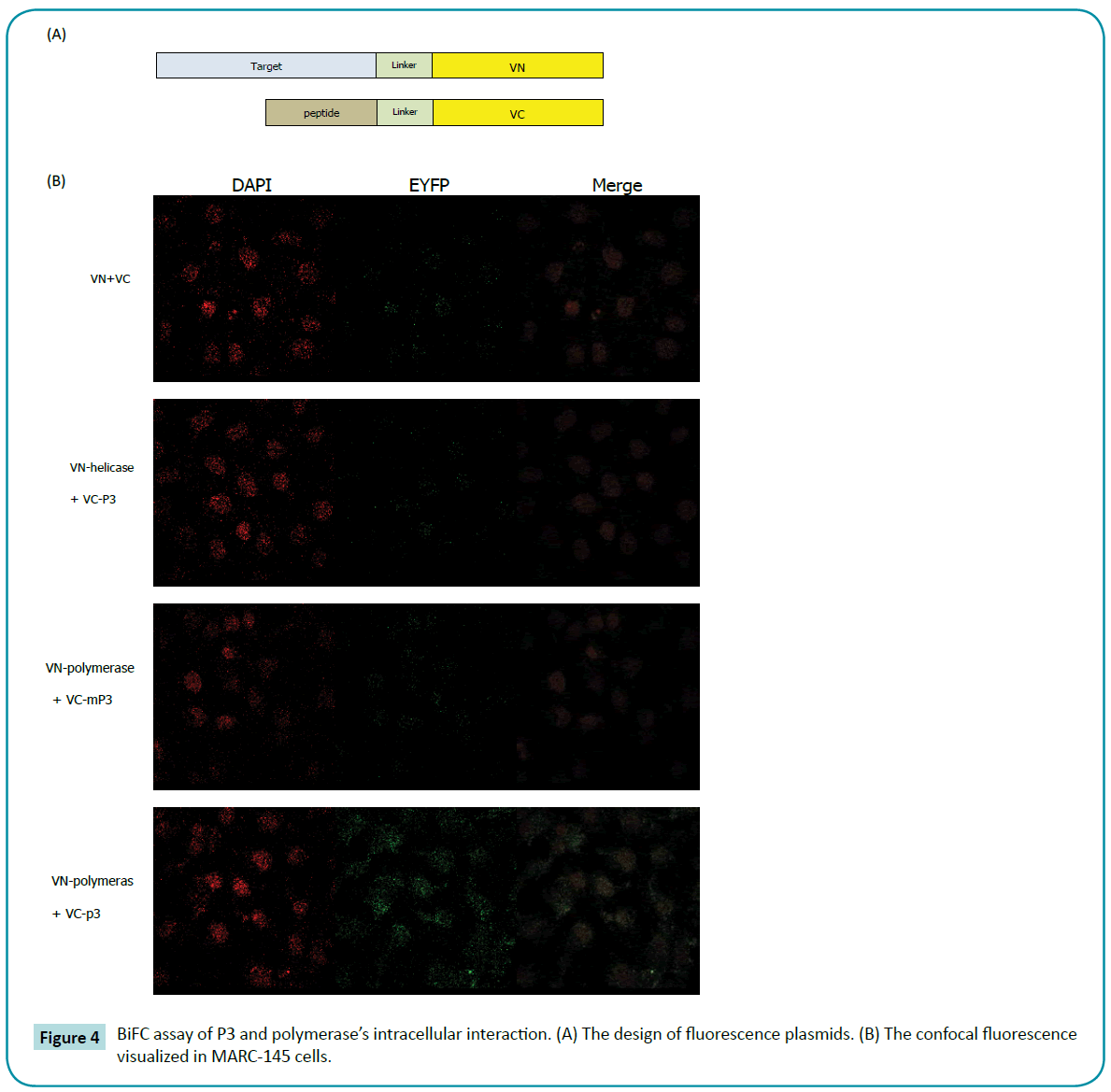
Figure 4: BiFC assay of P3 and polymerase’s intracellular interaction. (A) The design of fluorescence plasmids. (B) The confocal fluorescence visualized in MARC-145 cells.
Antiviral activity of combined peptides
In previous study, we find P9, APP-1, P3 and APP-3 showed highest antiviral activity in vitro. To determine the potentially combined antiviral effect of these peptides, two, three or four peptides were used in antiviral experiment. We found that P3 combined with p9 exerted synergistic antiviral activity against PRRSV in vitro (Figure 5).
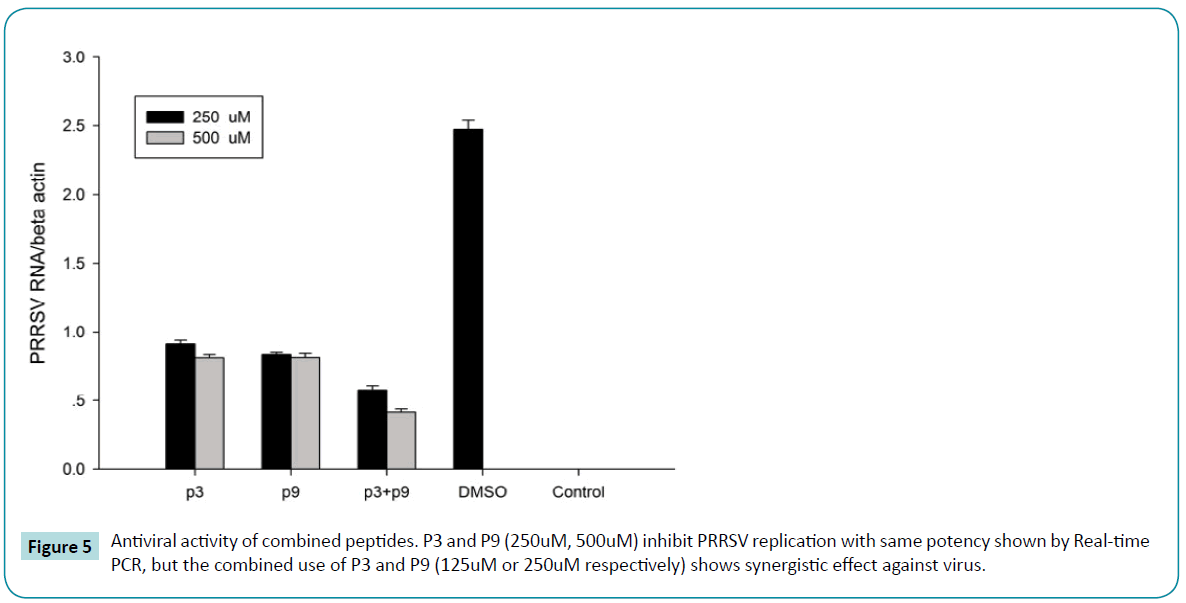
Figure 5: Antiviral activity of combined peptides. P3 and P9 (250uM, 500uM) inhibit PRRSV replication with same potency shown by Real-time PCR, but the combined use of P3 and P9 (125uM or 250uM respectively) shows synergistic effect against virus.
Discussion
In recent years, PRRSV infection has seriously harmed the pig industry. PRRSV infection has caused great economic loss, especially in the years 2005-2008 [2,6]. To date, there is no effective drug that can control PRRSV infection, and PRRSV vaccination is ineffectively. To investigate the novel strategy against PRRSV infection, in this study, the key protein involved in PRRSV replication was expressed and its high-affinity peptides were screened following phages display. One of selected peptide P3 showed very high extracorporeal antiviral activity against PRRSV replication.
PRRSV polymerase is an RNA-dependent RNA polymerase and is an enzyme specific to viruses. Therefore, peptides screened by phages could play a role in the inhibition of this specific virus enzyme in cells without affecting cell enzyme activity [10,14,26]. Several peptides were obtained from phage display in previous study. Based on antiviral test, it was found that the antiviral ability of P3 was high. P3’s pI value was 12.48, which indicated some characters like cationic anti-microbial peptides with antiviral activity. This pI value got close to some animal cathelicidin. The cationic character of antimicrobial peptides facilitated them to get closer to target; it is an important character to anti-microbial peptides. Our uptake experiments showed P3 could be effectively absorbed by cell.
Peptide inhibitors can be degraded and metabolized by intracellular proteases after they enter the cell [27,28]. Stability in cells is an important index to evaluate antiviral peptide [29,30]. After invaded cells, viruses persist and form new virus particles, which results in transmission of infection from one cell to another. This process requires that virus inhibitors exist stably in cells for a prolonged time [29]. Our study detected and observed the timecourse of P3 peptide in infected cells. Our results also indicated that P3 played an inhibitory role in cells in a dose-dependent manner. Virus concentration in cells increased with time, but when compared with the PBS control group, the quantity of virus and speed of virus were both lower in the presence of P3 [18].
We further tested the inhibitory effect of P3 added at 4 h before and after virus infection, which represented the virus prevention and the treatment effects of P3 [31,32]. The results showed that P3 added before cell infection had higher antiviral ability than P3 added at 0 h or 4 h after infection. In the +4 h group, P3 was added at 4 h after virus infection, and we calculated that P3 entered the cells probably at 5-6 h. In this 5-6 h period, viruses had enough time to complete intracellular capsid removal and polymerase expression [29,33-35]. Based on these results, we suggest that P3 plays an inhibitory role, and its inhibition efficiency was comparatively much lower than that of the –4 h group and 0 h group.
Our BiFC experiment showed intensively interaction between P3 and PRRSV polymerase. These results proofed that P3 is interacting with polymerase intracellular but not others [25,36]. Our studies show that the PRRSV Polymerase is an attractive drug target. Furthermore, we found that P3 is potent antiviral peptide with low cytotoxic at different concentrations. Thus, further development of P3 as an antiviral agent may lead to a new type of polymerase inhibitors that is expected to inhibit PRRSV.
Acknowledgments
This study was supported by grants from the National Basic Research Program, China (973 Plan, grant no. 2014CB542702).
8454
References
- Meulenberg JJ, Hulst MM, Meijer DE, Moonen EJ, Den BPL, et al. (1993) Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology 192:62–72.
- Mateu E, Diaz I (2008)The challenge of PRRS immunology. Vet J 177:345-351.
- Botner A, Strandbygaard B, Soensen KJ, Have, Madsen KG, et al. (1997) Appearance of acute PRRS-like symptoms in sow herds after vaccination witha modified live PRRS vaccine. Veterinary Record 141:497-499.
- Christopher-Hennings J, Nelson EA, Hines RJ, Nelson JK, Swenson SL, et al, (1995). Persistence of Porcine Reproductive and Respiratory Syndrome Virus in Serum and Semen of Adult Boars. J Vet Diagn Invest 7: 456-64.
- Madsen KG, Hansen CM, Madsen ES, Strandbygaard B, Botner A, Soensen KJ (1998). Sequence analysis of porcine reproductive and respiratory syndrome virus of the American type collected from Danish swine herds. Archives of Virology 143:1683-1700.
- Storgaard T, Oleksiewicz MB, Botner A (1999) Examination of the selective pressures on a live PRRS vaccine virus. Arch Virol 144: 2389-2401.
- Meng XJ, Paul PS, Halbur PG, Lum MA (1995) Phylogenetic analyses of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the USA and Europe. Arch Virol 140:745–55.
- Mounir S, Mardassi H, Dea S, (1995) Identification and characterization of the porcine reproductive and respiratory virus ORFs 7, 5 and 4 products. AdvExp Med Biol 380:317-320.
- Nelson EA, Christopher-Hennings J, Benfield DA (1995) Structural proteins of porcine reproductive and respiratory syndrome virus (PRRSV). Advances in Experimental Medicine and Biology 380: 321-323.
- Fang Y, Kim DY, Ropp S, Steen P, Nelson EA et al. (2004) Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Research 100:229-235.
- Cafruny WA, Duman RG, Wong GHW, Said S, et al. (2006) Porcine reproductive and respiratory syndrome virus (PRRSV) infection spreads by cell-to-cell transfer in cultured MARC-145 cells, is dependent on an intact cytoskeleton, and is suppressed by drug-targeting of cell permissiveness to virus infection. Virology Journal 3:90.
- Bai F, Town T, Pradhan D, Cox J, Ashish, et al. (2007) Antiviral peptides targeting the west nile virus envelope protein. J Virol 81: 2047-2055.
- Bajorath J, Peltason L, Wawer M, Guha R, Lajiness MS, et al. (2009) Navigating structure-activity landscapes. Drug Discov Today 14: 698-705.
- Hall PR, Hjelle B, Njus H, Ye C, Bondu-Hawkins V, et al. (2009) Phage display selection of cyclic peptides that inhibit Andes virus infection. J Virol 83: 8965-8969.
- Hancock RE, Sahl HG (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24: 1551-1557.
- Ho KL, Yusoff K, Seow HF, Tan WS (2003) Selection of high affinity ligands to hepatitis B core antigen from a phage-displayed cyclic peptide library. J Med Virol 69: 27-32.
- Peng B, Chen H, Tan Y, Jin M, Guo A (2006). Identification of one peptide which inhibited infectivity of avian infectious bronchitis virus in vitro. Sci China C Life Sci 49: 158-163.
- Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W (2006) Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev 58: 1622-1654.
- Xu H, Wang J, Yu Z, Lv F, Hou J (2011) [Screening of polypeptides binding to porcine reproductive and respiratory syndrome virus by phage display library]. Wei Sheng Wu XueBao 51: 127-133.
- Xiaofeng R, Mingcui W, Jiechao Y, Guangxing L (2010) Phages Harboring Specific Peptides That Recognize the N Protein of the Porcine Reproductive and Respiratory Syndrome Virus Distinguish the Virus from Other Viruses. JClinic Microbiol 48:1875-1881.
- Liu K, Feng X, Ma Z, Luo C, Zhou B, et al. (2012) Antiviral activity of phage display selected peptides against Porcine reproductive and respiratory syndrome virus in vitro. Virology 432:73-80.
- Oleksiewicz MB, Botner A, Madsen KG, Storgaard T (1998) Sensitive detection and typing of porcine reproductive and respiratory syndrome virus by RT-PCR amplification of whole viral genes. Vet Microbiol 64:7-22.
- SachsL A, Schnurr D, Yagi S, Lachowicz-Scroggins ME, Widdicombe JH (2011) Quantitative real-time PCR for rhinovirus, and its use in determining the relationship between TCID50 and the number of viral particles. Journal of Virological Methods 171:212-218.
- Shimozono S, Miyawaki A (2008) Engineering FRET constructs using CFP and YFP. Methods Cell Biol 85: 381-393.
- Shyu JY, Liu H, Deng X, Hu CD (2006). Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 40:61-66.
- Hagiwara K, Kondoh Y, Ueda A, Yamada K, Goto H, Watanabe T (2010) Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo-cross-linked chemical arrays. BiochemBiophys Res Commun 394:721-727.
- Hinson ER, Cresswell P (2009) The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic-helix. Proc Natl AcadSci U S A106:20452-20457.
- Wang H, Ooi EV, Ang PO Jr (2008) Antiviral activities of extracts from Hong Kong seaweeds. J Zhejiang UnivSci B 9: 969-976.
- Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, et al. (2010)A broad-spectrum antiviral targeting entry of enveloped viruses. Proceedings of the National Academy of Sciences 107:3157-62.
- Yan R, Zhao Z, He Y, Wu L, Cai D, et al. (2011) A new natural α-helical peptide from the venom of the scorpion Heterometruspetersii kills HCV. Peptides 32: 11-19.
- Konduru K, Kaplan GG (2010) Determinants in 3Dpol modulate the rate of growth of hepatitis A virus. J Virol 84: 8342-8347.
- Münch J, Ständker L, Adermann K, Schulz A, Schindler M, et al. (2007) Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 129: 263-275.
- Castel G, Chtéoui M, Heyd B, Tordo N (2011) Phage display of combinatorial peptide libraries: application to antiviral research. Molecules 16: 3499-3518.
- Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland et al. (2008) A virocidal amphipathic-helical peptide that inhibits hepatitis C virus infection in vitro. Proc Natl AcadSci U S A105:3088-3093.
- Li Q, Zhao Z, Zhou D, Chen Y, Hong W, et al. (2011) Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 32: 1518-1525.
- You JH, Howell G, Pattnaik AK, Osorio FA, Hiscox JA (2008) A model for the dynamic nuclear/nucleolar/cytoplasmic trafficking of the porcine reproductive and respiratory syndrome virus (PRRSV) nucleocapsid protein based on live cell imaging. Virology 378:34-47.