Keywords
Prion; Diagnostics; TSEs; PrPSc; PrPc; Transmissible Spongiform Encephalopathies
Introduction
Prion diseases are rare but deadly conditions of central nervous system, manifesting neurodegenerative symptoms such as convulsions, dementia, ataxia and behavioral changes. Because of their transmissibility feature and the characteristic sponge-like microscopic appearance in the affected brain tissue, they are also called Transmissible Spongiform Encephalopathies (TSEs). The incubation period for prion disease is long, but once the clinical signs appear, the disease progresses fast and rapidly results in death. Although symptom relief treatments exist, no survival benefit-conferring drug is available for the treatment of human prion diseases [1,2]. The variant Creutzfeldt-Jakob disease (vCJD) crisis among humans in 1996 in The United Kingdom was caused by the consumption of Bovine Spongiform Encephalopathy (BSE) infected cattle, testifying that the potentially prevalent prion diseases could spread from food animals to human beings, and therefore the crisis raised deep concerns about human health and economy [3]. It is also dangerous to omit those infected people without significant clinical signs, because the time from being infected to showing signs of the disease can be up to 50 years [4], and during this period of incubation time the infected people can be pathogen carriers and may spread the prion diseases through medical/dental instrument contamination, transfusion or transplantation [5]. Therefore, it is important to develop sensitive techniques for the diagnosis of prion diseases not only for the patients’ symptom relief per se, but also for the protection of public health. Traditionally, prion diseases were usually diagnosed by clinical signs and confirmed by post-mortem examination, but there was no sufficient method for early stage diagnosis of the diseases. Recently, the newly improved in vitro amplification diagnosis methods may serve as a promising solution to this shortage, and may provide the potential noninvasive screening tools for disease surveillance. In this review, we give a brief overview of the histories of prion diseases’ discovery, pathogenesis and the molecular diagnosis, and in particular we summarized the fast and accurate noninvasive screening tests developed recently, in order to shed some light on the early diagnosis and surveillance of prion diseases.
Discovery History of Prion Diseases
In human beings, there are 6 phenotypes of prion diseases: kuru disease, Creutzfeldt-Jakob disease (CJD), Gerstmann- Sträussler-Scheinker syndrome (GSS), Fatal Familial Insomnia (FFI), variant Creutzfeldt-Jakob disease (vCJD) and variably protease sensitive prionopathy (VPSPr); In herbivorous ungulate animals, there is scrapie in sheep and goats, Bovine Spongiform Encephalopathy (BSE, also known as "mad cow disease") in cattle, Chronic Wasting Disease (CWD) in deer and elk; In carnivorous animals, there is Transmissible Mink Encephalopathy (TME) in mink, and Feline Spongiform Encephalopathy (FSE) in cat and cheetah. It is very likely that more species carry this type of disease. All these prion diseases share the common features including the fatal neurodegenerative process, sponge-like brain, and disease transmission ability. Figure 1 shows the timeline of the discovery of the known prion diseases, and we summarized here the brief history of each type of them, from the first discovery to the well-established stage typified by the successful inoculation to model animals.
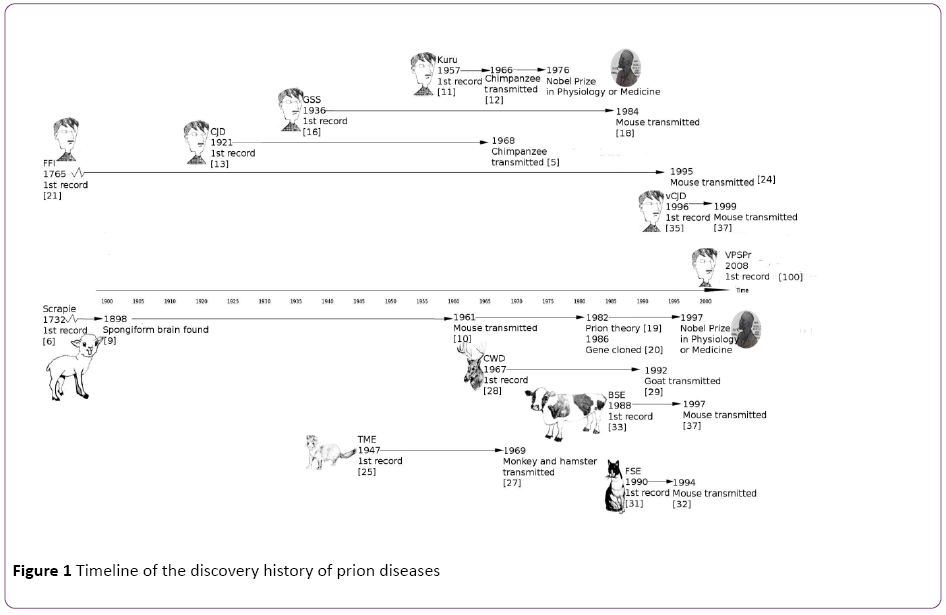
Figure 1: Timeline of the discovery history of prion diseases
Scrapie in sheep and goat
Scrapie is the earliest prion disease described. The first record dates back to 1732, when sheep scrapie was described in Great Britain [6]. Scrapie can infect both sheep and goats, spread among them through contact, and cause infected animals scraping off wool or hair, losing weight, and displaying odd behaviors [7,8]. In 1898, the brain lesion in the scrapieinfected sheep was noticed, which is the first mention of the characteristics of the spongiform encephalopathy [9]. In 1961, Chandler discovered that scrapie could be given to mice, which accelerated the progress of scrapie research [10], and the method of transmission to a model animal was widely used on other prion disease studies since then. After the researchers found diseases with clinical signs due to the spongiform encephalopathy, they would study the diseases in model animals, which thereby quickened the pathogenesis research.
Kuru, CJD, GSS, FFI and VPSPr in human beings
In 1957, Gajdusek reported kuru disease in the Fore tribe located in Papua, New Guinea [11]. The disease was named after “kuru” because this local word translates to “trembling from fear or cold”, well reflecting the unique tremor characteristics of the disease during the early stages. Gajdusek later determined the cause of kuru as the ritualistic cannibalism in Fore tribe after he achieved the transmission of kuru to chimpanzees [12].
In 1920s, Creutzfeldt and Jakob found another fatal disease of spongiform encephalopathy, Creutzfeldt-Jakob disease (CJD), which leads to late-onset neurodegenerative dementia with a 6-months overall survival rate of 30% [13]. In 1968, Gibbs did the CJD transmission to the chimpanzee [5]. By 1972, Gajdusek and other researchers realized that scrapie, kuru and Creutzfeldt-Jakob diseases were all transmissible subacute spongiform encephalopathies, and they proposed that these diseases were caused by a slow virus [14]. In 1978, CJD was transmitted to the mouse model by Manuelidis [15].
GSS was firstly reported in 1936 [16], and was eventually found to share similar pathogenic process with CJD [17] and can be transmitted to rodent models [18]. In 1982, Prusiner found that prion was the actual cause of scrapie [19], and he later categorized the three degenerative human diseases similar to scrapie, kuru, CJD and GSS, as prion diseases [20]. Prusiner was awarded with the Nobel Prize in 1997 in Physiology or Medicine for his discovery. FFI can be dated to 1765, when the first recorded death of a patient who had suffered from insomnia for more than a year [21]; however, it was identified in 1982 [22] and was included into the prion disease category [23]. FFI was conformed transmissible to laboratory animals in 1995 [24].
VPSPr was first reported in 2008 [25]. It is a sporadic human prion disease that is heterogeneous in terms of protease sensitivity [26]. VASPr is not a well-studied prion disease yet and there are no published studies of its transmissibility to animal models.
TME in mink, CWD in deer and elk, and FSE in cats
The first observations of transmissible mink encephalopathy were reported in 1947 [27,28]. Transmissible mink encephalopathy is a progressive, fatal disease of ranch mink. The disease has been experimentally transmitted to monkeys and hamsters [29]. In 1967, Chronic Wasting Disease (CWD) in North American cervids was first observed, and was found transmissible by intracerebral inoculation to cattle, sheep, and goats [30]. Surveillance efforts for CWD are more difficult, because the disease was not only found in captive cervids [31], but also in free-ranging cervids [32].
In 1990FSE was firstly found in a cat, with a spongiform encephalopathy characterized principally by grey matter neuropil spongiosis [33], and this disease was transmitted to mice later [34]. More cases of FSE were found in captive wild cats including cheetahs, pumas, ocelots, tigers, lions and Asian golden cats [35].
BSE in cow and vCJD in human
BSE and vCJD are introduced together since they are closely related. vCJD was found in human beings as a result of transmission of bovine prions from BSE-infected cattle to human. It was in 1980s that a British cow was diagnosed with BSE, also known as "mad cow disease". At the beginning, the spreading of BSE in British cattle raised the fear of transmission to humans, since people could accidentally consume BSE infected cows that carry the transmissible agent prion [36]. Unfortunately, the fears turned to become reality in 1990s when the variant Creutzfeldt–Jakob Disease (vCJD) found in humans was confirmed as a result of transmission of bovine prions from BSE-infected cattle [37-39]. vCJD was identified as the consequence of eating diseased cattle, and it tends to occur in younger people. vCJD causes prominent psychiatric or sensory symptoms and delayed the onset of neurological abnormalities, which are different with other CJDs [40].
Pathogenesis of Prion Diseases
Early studies on the unconventional “viruses” and the success on stopping kuru
The observation on scrapie showed that birthing fluids and placenta from infected females contained scrapie agent and caused disease transmission to healthy newborn lambs. Those newborn lambs that shared the same lambing pen were extremely susceptible to infection. Back then, the known causes of transmissible diseases were limited to certain bacterial, viral, fungal, or parasitic agents. Wilson started to postulate that the agent was virus [41]. Many early scrapie researchers agreed because they found the agents of scrapie were very small, which was consistent with the postulation of virus [42]. Gajdusek in his work on kuru also proposed that the kuru agent should be a kind of transmissible virus [43]. However, Alper noticed that ultra-violet irradiation of suspensions of infected mouse brain extracts did not affect the infection of the extracts to other mice, therefore concluded that the agent probably did not depend on nucleic acids for its ability to replicate since UV could have killed the virus [44]. When Gajdusek transmitted the kuru, CJD and scrapie diseases to squirrel monkeys, he assumed that the agents of the 3 diseases were all “slow virus”, which means the virus might slowly develop from very residual amount that survived the environmental damaging factors [45]. Despite the fact that the suspected slow virus had never been identified, after the stopping of cannibalism in 1960, kuru cases decreased and finally had been completely eradicated. Because of his contribution on kuru research, Gajdusek won the 1976’s Nobel Prize in Physiology or Medicine.
Finding out prion as the agent and the establishment of the prion theory
In 1982, after a series of studies of the characteristics of the agents of these diseases, Prusiner found evidences to indicate the diseases’ agent is associated with a protein [46,47]. He termed a proteinaceous infectious agent, “Prion”, to explain the disease pathogenesis [48]. There are many debates against prion theory in the scientific community from the beginning [49], to the time after Prusiner was awarded the Nobel Prize in Physiology or Medicine for his discovery of prion in 1997, and even to very recent days [50-52]. However, prion theory was eventually well established and based on this theory, a new class of infectious diseases, “prion diseases”, had been categorized.
Based on this theory, the diseases are transmitted by a proteinaceous infectious particle, prion. On the molecular level, as shown in Figure 2, prion is the infectious abnormal conformation isoform (PrPSc, PrP means prion protein, and Sc was named after scrapie) of a normal cellular prion protein (PrPc), while PrPc is primarily located on the cell surface of neurons within the central nervous system but its specific function is not completely understood. PrPSc forms amyloids and is infectious because when touch PrPc, it converts PrPc into PrPSc [53]. The accumulation of PrPSc seriously damage cells, cause gliosis and neuronal loss. Furthermore, PrPSc aggregates to make plaques known as amyloids and form vacuoles in the neurons, which cause the typical spongiform structure in the infected brain [54]. Since prion unit is a protein molecular, even electron microscope can only show the amyloids but not individual prions, which explained why the agents were deduced to be extremely small before. Prion is protein-only, which explained why the agents were resistant to procedures that usually break down nucleic acid and destroy biological forms of other pathogens. Because prion is an abnormal form of a normal genetically encoded protein, it causes the genetic feature of the familial prion diseases, and doesn’t cause immune response in the host.
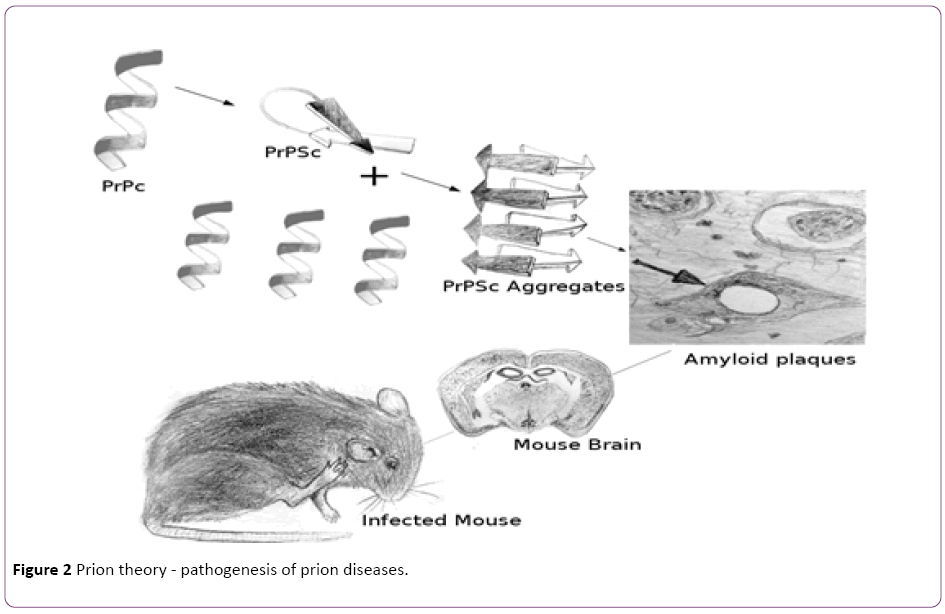
Figure 2: Prion theory - pathogenesis of prion diseases.
It has been reported that transmissible prion like protein agent from a filamentous fungus can be generated in vitro [55], demonstrating the validity of the protein-only hypothesis. Although the prion based pathogenic mechanisms were first described in mammals, now they may have wider relevance since similar proteinaceous infectious particles were also found in fungi and yeast [56].
A PrPc shadow protein called Shadoo was found to have protective properties [57] to the disease, and the downregulation of PrPc was an element of disease pathogenesis [58]. Recently, there are some inhibitors to the formation of PrPSc were found, raising hope of beneficial treatments of prion diseases [59,60]. Recent findings also suggested that the misfolding of prion enhance the formation of p-tau and aggregation of α-synuclein and ubiquitin, which might lead to therapeutic modalities [61]. Prion disease vaccine possibility based on the significant progress on antibodies identifying PrPSc-specific epitope was also discussed [62,63]. A recent study found that heparanase plays a protective role in prion disease infection and progression by interfering the heparan sulfate involved in the pathogenesis of prion disease [64]. This in vivo study showed that the scrapie onset and progression were dramatically delayed in scrapie infected transgenic mice over-expressing heparanase, and thus revealed that heparanase is another potential treatment target. However, the basic mechanism on how PrPSc captures and recruits PrPc and converts it into PrPSc remains unclear till now. The discovery of the mechanism in this procedure not only may lead to the development of new diagnostic and therapeutic approaches, but also may construct another milestone of basic biology science and culture the third Nobel Prize winner on prion diseases.
Prpsc Detection Tools for Prion Disease Diagnosis
Prion diseases are traditionally diagnosed with clinical signs, electroencephalography and magnetic resonance imaging, and confirmed by post-mortem pathological examination. Molecular diagnosis generally can reach higher level of sensitivity and specificity, which are important for both diagnosis confirmation and preclinical screening. Since PrPSc is the agent of prion diseases, it becomes the only reliable molecular marker of prion diseases. There were molecular markers for prion disease, such as 14-3-3 protein, S100 protein and neuron-specific enolase (NSE), but those markers were not as good as PrPSc with respect to specificity. PrPSc-based diagnosis is not only the final pathological conformation test of the diseases, but also the first candidate for the preclinical screening [65,66]. In Table 1, we listed the classical PrPSc detecting methods, which are grouped into 3 categories, including in vivo amplification category, non-amplification category and in vitro amplification category. The in vivo amplification method is to use bioassay in laboratory animals. It is sensitive and reliable but time consuming (months or years), and sometimes hard to overcome species barriers. The non-amplification methods have good specificity and they are traditionally used in clinical diagnosis, but in most cases postmortemly. Only when some of them use enhanced techniques like capture enrichment and florescence, they can have high sensitivity for ante-mortem blood test. The in vitro amplification methods are sensitive enough to detect misfolded prion proteins in the asymptomatic stages, even in the noninvasive body fluid like urine.
| Category |
Method |
Mechanism |
Reference |
If detects Proteinase K-sensitive PrPSc? |
Target Specimens |
Notes |
Commercial Kits |
| In vivo Amplification |
Animal BioAssay |
Measures ID50 or LD50 in inoculated animals |
Chandler[10] |
Yes |
Tissue, blood, urine, saliva |
Amplified signs, but costly and sometimes has species barriers. |
None |
| Non Amplification |
Western Blot |
PrP antibody stain |
McKinley [67] |
No |
Tissue |
Can detect strain types. |
Prionics-Check Western |
| |
ELISA |
PrP antibody stain |
Barry [68] |
No |
Tissue, blood |
Convenient for antigen capture enrichment. |
Prionics Check, Enfer TSE,Bio-Rad TeSeE,Roboscreen Beta Prion BSE, Institut Pourquier Speed’it BSE, Roche PrionScreen, IDEXX HerdChek BSE |
| |
IHC |
PrP antibody stain |
McBride [69] |
No |
Tissue |
Can show locations in the tissue, but may miss antigen inactivated PrPSc. |
Ventana IHC Systems |
| |
CDI |
Conformation dependent immunoassay using epitope protection |
Safar [70] |
Yes |
Tissue |
Can detect strain types. |
InPro CDI-5, Ceditect BSE |
| |
DELFIA |
Dissociation-Enhanced Lanthanide Fluorescent Immunoassay, measures percentage of insoluble PrPSc |
Barnard [71] |
Yes |
Tissue, Blood |
Integrated time-resolved fluorescence signal. |
PE DELFIA TRF |
| |
MPD |
misfolded protein diagnostic, attached excimer fires when the peptide converts to ß-sheet conformation |
Grosset [72] |
Yes |
Tissue, Blood |
Simple steps without using any antibody. |
None |
| |
SOFIA |
Surround optical fiber immunoassay |
Chang [73] |
Yes |
Tissue,Blood,Urine |
Cooperating multiple antibodies. |
None |
| In vitro Amplification |
Cell BioAssay |
Detects PrPSc in inoculated cells |
Bosque [74] |
No |
Tissue, Blood |
Amplified signal, sometimes has species barriers. |
None |
| |
PMCA |
Protein misfolding cyclic amplification |
Saborio [75] |
Yes |
Tissue,Blood,CSF,Urine |
Amplified signal. |
None |
| |
ASA |
Amyloid seeding assay, uses amyloid dye ThT |
Colby [76] |
Yes |
Tissue,Blood,CSF |
Amplified signal, low background, needs precipitation step in seed preparation. |
None |
| |
RT-QUIC |
Real-time quaking-induced conversion, uses amyloid dye ThT |
Atarashi [77] |
Yes |
Tissue,Blood,Nasal Swab |
Amplified signal, has simple shaking step and low background. |
None |
Table 1: PrPSc-based diagnosis methods
In vivo amplification detection of PrPSc
Bioassays that involve the use of laboratory animals are often used for the detection and titration of PrPSc, because PrPSc can convert PrPc to PrPSc in vivo and thus cause observable clinical signs and death. The titer can be measured by the incubation period in the experimental animals inoculated with serial dilutions of the test material [78], or determined by the median infective or lethal dose (ID50, LD50), which is the dose of prion that causes infection or fatal situation in 50% of the inoculated animals respectively [79]. Although they are extremely sensitive that can detect PrPSc in blood [80,81], urine and saliva [67], they usually require long time of incubation and sometimes have species barrier problem. Transgenic mouse for the test is helpful to break through the species barrier and decrease the incubation time, but the cost is still high on time and money.
Non-amplification detection of PrPSc
Traditional immunological detections: According to the fact that parts of PrPSc cannot be digested by proteinase, immunological detections of the proteinase K-resistant part of PrP were developed, including traditional Western blot [67], Immunohistochemistry (IHC) [69] and enzyme-linked immunosorbent assay (ELISA) [68]. IHC can show microscopic location of PrPSc but sometimes has antigen inactivation problems in the fixed tissues, so that antigen retrieval is needed in IHC [82]. Western blots can detect 1,000-fold dilution of scrapie-infected brain homogenate, while ELISA is more rapid than Western blot and is more convenient for antigen capturing enrichment.
ELISA can increase sensitivity significantly after capture enrichment so that it detect PrPSc in blood samples [83,84]. There is only one available commercial Western Blot kit, Prionics’ Check Western, while there are several commercial ELISA kits for screening large numbers of animals, such as Prionics’ Check serial, Enfer’s TSEBio-Rad’s TeSeE, Roboscreen Beta’s Prion BSE, Institut Pourquier’s Speed’it BSE, and Roche’s PrionScreen.
Not all of the PrPSc were proteinase resistant [84,85], so the post-proteinase detection method is not sufficient for the proteinase sensitive PrPSc. Therefore, ELISA methods rely on antibodies or chemical media that specifically bind to PrPSc were used to detect proteinase K sensitive PrPSc [86,87]. The first commercial test based on PrPSc specific recognition is IDEXX HerdChek BSE Antigen EIA, which uses a polymeric ligand developed to capture PrPSc specifically [88].
Conformation-dependent immunoassay (CDI): Studies found that the immunoreactivity of PrPSc is greatly enhanced by guanidinium thiocyanate denaturation [89], because the conformation of PrPSc changed and exposed the hidden epitopes during the guanidinium thiocyanate denaturation [90]. CDI uses the ratio of antibody binding to denatured/ native PrP as a measure of the concentration of PrPSc, and tells the different strains according to the different positions on the plotted denatured/native PrP ratio graph [70]. The enhanced CDI can detect PrPSc in bodily fluids of CJD/vCJD patients [91].
Dissociation-Enhanced Lanthanide Fluorescent Immunoassay (DELFIA): This method uses a two-site immunoassay. It determines the PrP concentrations of two extracts processed with two concentrations of guanidine hydrochloride, and then measure the percentage of insoluble PrPSc as the diagnostic parameter. The assay is precise because it uses the very robust and sensitive time-resolved fluorescence kit that is commercially available from PE life science [71].
Misfolded protein diagnostic (MPD): MPD assay is a simple detection method based on the conversion of the synthetic prion peptide in the presence of PrPSc. The signal is generated from excimer formation of the fluorophore attached to the peptide. When the PrPSc in the sample convert the artificial peptide to β-sheet, the two attached fluorophore form an excimeric dimer to emit fluorescence [72].
Surround optical fiber immunoassay (SOFIA): In prion antibodies, there is a positive immunocooperativity phenomenon, which means that after one PrPSc specific monoclonal antibody binds to PrPSc, other PrPSc epitopes start to open for the other specific monoclonal antibody. Based on this phenomenon, SOFIA detection was developed. It consists an immunocapture step and a highly sensitive fiber optical-based laser-induced fluorescence detection step [73]. The high sensitivity of this assay is a good feature for preclinical PrPSc detection in biological fluids [92].
In vitro amplification detection of PrPSc: Since PrPSc is a misfolding form of a protein and it has no specific maker on gene level, it cannot be detected through the traditional sensitive PCR-based methods. However, there are in vitro systems for the amplification of PrPSc, conceptually analogous to PCR cycling, to increase the detection sensitivity. They rely on imparting very small amounts of misfolded protein seeds and amplifying them into larger amyloidogenic or proteinaseresistant protein aggregates.
Cell bioassay: Animal bioassay is a sensitive and reliable detection method based on in vivo PrPSc amplification, but it is expensive and time-consuming. High PrPSc infectivity cell lines provided good ex vivo models for PrPSc detection [74,93]. The cell-based bioassays usually use traditional Westen blot and ELISA to detect the amplified PrPSc in the cell lines, and they had shorter incubation time while showed similar sensitivity with animal bioassay [94]. Like in the animal bioassays, there are species barriers in cell bioassays, but the overexpression of PrPc in the cell lines may overcome the species barriers [95,96]. Cell bioassay is an efficient method when the suitable immortalized cell line with certain DNA content is available.
Protein misfolding cyclic amplification (PMCA): PMCA uses in vitro cyclic amplification system that allows a rapid conversion of large excess recombinant PrPc into a proteinaseresistant PrPSc-like form in the presence of PrPSc template from the samples, and then detects the proteinase-resistant PrPSc by Western blot. In every cycle, converted PrPSc aggregate when the template PrPSc incubate with PrPc, and then the aggregates are disrupted by sonication to produce multiple PrPSc templates for the next cycle of amplification [75]. This method is sensitive enough for PrPSc detection in blood in the asymptomatic stages of prion diseases [97-100].
Amyloid seeding assay (ASA): Because PrPSc can convert PrPc into PrPSc and then cause amyloid formation, ASA uses PrPSc as seed to convert PrPc into amyloids, and then detects the amyloids by a fluorescence shift in the amyloid specific dye Thioflavin T [76]. This ASA reaction is an in vitro amplification system for PrPSc detection, and the low background fluorescence in the absence of amyloid helps to increase the sensitivity, which seems high enough to do the pre-clinical test. However the impurities in the sample tended to inhibit the amyloid formation, so ASA used Precipitation with phosphotungstic acid (PTA) [101] to partially purify the seed in the sample preparation.
Real-time quaking-induced conversion (RT-QUIC): A quaking-induced conversion (QUIC) was developed to be in vitro amplification system similar to PMCA, but with a constant shaking incubation to replace the sonication cycles, which has the problem of varied delivery of vibrational energy to the samples [102]. There is an enhanced QUIC method in which immunoprecipitation is applied before the standard QUIC [103]. RT-QUIC is a method that combines QUIC with thioflavin T (ThT) fluorescence dye that is used in ASA, to do a real-time amyloid formation test [77]. RT-QUIC is sensitive enough for body fluid detection, and it can be used in noninvasive screening [104].
Discussion
Since prion diseases are intra-species and inter-species transmissible, they are potentially prevalent in human through people’s food consumption and iatrogenicity. Except for the majority of sporadic and genetic human prion disease cases, a few historic cases showed that their diseases were environmentally acquired through cannibalism, infected food animal consumption, and iatrogenicity. The cannibalism caused spreading was successfully terminated when the cannibalism was stopped. However, the more recent variant Creutzfeldt-Jakob disease (vCJD) crisis caused by consumption of bovine spongiform encephalopathy (BSE) infected cows, still reminds us that the infected food animals is a big threat to people. Prion infected patients without significant clinical signs remain as another threat, because the diseases may incubate in people’s body for a long latent period and spread through medical or dental instrument contamination, transfusion or transplantation. Some sensitive and accurate prion disease diagnosis tools are very important not only for the early intervention for the patients’ symptom relief, but also for the protection of public health against disease prevalence. Although currently the prion disease diagnosis tools are not sufficient for the clinical use or pre-clinical surveillance use, the recent advances of PrPSc detection technique provide the potential screening tools for the early diagnosis and disease surveillance.
There are mutual benefits between diagnosis studies and mechanism studies. Even before the establishment of the prion theory, the early diagnosis studies had revealed the characteristics of the disease and had suggested prevention approaches. For example, in early studies, the animal bioassay developed from scrapie diagnosis studies directly led to the determination of other prion diseases, and also consequently led to the termination of kuru disease. In later diagnosis studies after the setup of prion theory, some new findings, like the proteinase K insensitive PrPSc and positive immunocooperativity in the binding of the antibodies to epitopes, were very helpful for understanding the conformation differences in the prion strains. On the contrary, the prion theory discovered the pathogen behavior, and helped on identifying PrPSc as the most reliable marker for diagnosis of prion diseases. Based on PrPSc/PrPc transformation theory, more sensitive amplification detection methods were developed to diagnosis prion disease. Therefore, the recent breakthrough of the prion disease diagnostic studies, especially the newly modified methods that can detect body fluids, showed promising power to meet the clinical diagnosis needs. At the same time, the advanced diagnostic tools may also speed up the prion diseases pathogenesis studies, and even inspire therapeutic approaches.
In the diagnostic practice of prion diseases, including postmortem confirmation, clinical diagnosis and pre-clinical screening, the high specificity and sensitivity are always crucial. Usually, antibody recognition meets the need of specificity, especially when the different monoclonal antibodies are used for recognizing different epitopes, they can even tell the conformation differences in the prion strains. Only one method in our summarized method list, misfolded protein diagnostic method (MPD), does not use any antibody, but directly uses the recognition and binding ability between PrPSc and PrPc. As for increasing the sensitivity, capture enrichment in sample preparation, in vitro PrPSc amplification, and fluorescence application in the signal detection are the three main trends in prion diagnosis research. We introduced all the historical PrPSc detection methods even though some of them are not actively used, because an efficient way to improve the specificity and sensitivity in disease diagnosis is to combine the previous methods. The prion diagnosis history has showed that combinations often produce better output. For example RT-QUIC combined standard QUIC with ASA, and in enhanced QUIC, the standard QUIC was combined with the capture enrichment. There are many potential combinations in the current platforms such as combinations of CDI and DEFIA, PMCA and MPA, and so forth, which may turn out to be better diagnosis tools in future.
Since PrPSc is not evenly distributed and PrPSc involves different tissues in different prion diseases, it is critical to use the correct sampling strategy according to the development of a certain type of prion disease for the diagnostic tests. In the two recent non-invasive prion diagnosis studies in 2014, the RT-QuIC method found that the sensitivity of the olfactory mucosa–based test was higher than that of the cerebrospinal fluid–based test in the same sporadic CJD patients [103], while the PMCA method could only detected vCDJ in urine but not other human prion diseases [99]. Therefore, before the methods can be used diagnostically, further validation studies are needed to fully establish diagnostic sensitivities and specificities in clinical settings, such as tissue specific samples, pathogen types and strains, and stages of the prion diseases.
Acknowledgement
We are grateful to Dr. Jiangbing Zhou at Yale Neurosurgery department for the comments on this review. We also thank Ziting Wang for drawing all the cartoon elements in our figures.
8110
References
- Ghaemmaghami S, Russo M, Renslo AR (2014) Successes and challenges in phenotype-based lead discovery for prion diseases. J Med Chem 57: 6919-6929.
- Panegyres PK, Armari E (2013) Therapies for human prion diseases. Am J Neurodegener Dis 2: 176-186.
- Jasanoff S (1997) Civilization and madness: The great BSE scare of 1996. Public Understanding of Science 6: 221-223.
- Collinge J, Whitfield J, McKintosh E, Frosh A, Mead S, et al. (2008) A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Philos Trans R SocLond B BiolSci 363: 3725-3739.
- Gibbs CJ Jr, Gajdusek DC, Asher DM, Alpers MP, Beck E, et al. (1968) Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161: 388-389.
- Stockman S (1913) scrapie: an obscure disease of sheep. J Comp Pahol 26: 317-327.
- Brotherston JG, Renwick CC, Stamp JT, Zlotnik I, Pattison IH (1968) Spread and scrapie by contact to goats and sheep. J Comp Pathol 78: 9-17.
- Besnoit C, Morel C (1898) Note sur les lesions nerveuse de la tremblante du mouton. Rev Vet (Toulouse) 23: 397-400.
- CHANDLER RL (1961) Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1: 1378-1379.
- Gajdusek DC, ZigasV (1957) Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med 257: 974-978.
- Gajdusek DC, Gibbs CJ, Alpers M (1966) Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 209: 794-796.
- Lampert PW, Gajdusek DC, Gibbs CJ Jr (1972) Subacute spongiform virus encephalopathies. Scrapie, Kuru and Creutzfeldt-Jakob disease: a review. Am J Pathol 68: 626-652.
- Gibbs CJ Jr, Gajdusek DC (1972) Isolation and characterization of the subacute spongiform virus encephalopathies of man: kuru and Creutzfeldt-Jakob disease. J ClinPatholSuppl (R CollPathol) 6: 84-96.
- Manuelidis EE, Gorgacz EJ, Manuelidis L (1978) Transmission of Creutzfeldt-Jakob disease with scrapie-like syndromes to mice. Nature 271: 778-779.
- Gerstmann J, Straussler E, Scheinker I (1936) Ubereineeigenartigehereditar-familiareErkrankung des Zentralnervensystems. ZugleicheinBeitragzurFrage des vorzeitigenlokalenAlterns. Z Neurol 154: 736-762.
- Masters CL, Gajdusek DC, Gibbs CJ (1981) Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus of amyloid plaque deposition in the virus induced spongiform encephalopathies. Brain 104: 559-588.
- Tateishi J, Sato Y, Nagara H, Boellaard JW (1984) Experimental transmission of human subacute spongiform encephalopathy to small rodents. IV. Positive transmission from a typical case of Gerstmann-Sträussler-Scheinker's disease. ActaNeuropathol 64: 85-88.
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136-144.
- Prusiner SB (1985) Structure and biology of scrapie prions. Interrelationship Among Aging, Cancer and Differentiation. In: Pullman B, et al. (eds.), The Jerusalem Symposia on Quantum Chemistry and Biochemistry 18: 277-287.
- Max D T (2006) The family that couldn't sleep: a medical mystery. Random House New York Chapter I, page 3.
- Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, et al. (1986) Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 315: 997-1003.
- Gambetti P (1991) Fatal familial insomnia: a new human prion disease. Presented at the Prion Diseases in Animals and Humans Symposium, London.
- Collinge J, Palmer MS, Sidle KC, Gowland I, Medori R, et al. (1995) Transmission of fatal familial insomnia to laboratory animals. Lancet 346: 569-570.
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, et al. (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63: 697-708.
- Peden AH, Sarode DP, Mulholland CR (2014) The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. ActaNeuropatholCommun 2: 152.
- Hartsough GR, Burger D (1965) Encephalopathy of mink. I. Epizootiologic and clinical observations. J Infect Dis 115: 387-392.
- Liberski PP, Sikorska B, Guiroy D, Bessen RA (2009) Transmissible mink encephalopathy - review of the etiology of a rare prion disease. Folia Neuropathol 47: 195-204.
- Marsh RF, Burger D, Eckroade R, ZuRhein GM, Hanson RP (1969) A preliminary report on the experimental host range of the transmissible mink encephalopathy agent. J Infect Dis 120: 713-719.
- Williams ES, Young S (1980) Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16: 89-98.
- Williams ES, Young S (1982) Spongiform encephalopathy of Rocky Mountain elk. J Wildl Dis 18: 465-471.
- Wyatt JM, Pearson GR, Smerdon TN, Gruffydd-Jones TJ, Wells GA, et al. (1991) Naturally occurring scrapie-like spongiform encephalopathy in five domestic cats. Vet Rec 129: 233-236.
- Fraser H, Pearson GR, McConnell I, Bruce ME, Wyatt JM, et al. (1994) Transmission of feline spongiform encephalopathy to mice. Vet Rec 134: 449.
- Imran M, Mahmood S (2011) An overview of animal prion diseases. Virol J 8: 493.
- Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, et al. (1996) Transmission dynamics and epidemiology of BSE in British cattle. Nature 382: 779-788.
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, et al. (1996) A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347: 921-925.
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, et al. (1997) Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 389: 498-501.
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, et al. (1997) The same prion strain causes vCJD and BSE. Nature 389: 448-450, 526.
- Harris, David (Ed.) (2004) Mad Cow Disease and Related Spongiform Encephalopathies. Springer Science and Business Media page 126.
- WILSON DR, ANDERSON RD, SMITH W (1950) Studies in scrapie. J Comp Pathol 60: 267-282.
- Alper T, Haig DA, Clarke MC (1966) The exceptionally small size of the scrapie agent. BiochemBiophys Res Commun 22: 278-284.
- Gajdusek DC, Gibbs CJ (1964) Attempts to demonstrate a transmissible agent in kuru, amyotrophic lateral sclerosis, and other sub-acute and chronic nervous system degenerations of man. Nature 204: 257-259.
- Alper T, Cramp WA, Haig DA, Clarke MC (1967) Does the agent of scrapie replicate without nucleic acid? Nature 214: 764-766.
- Gibbs CJ, Amyx HL, Bacote A, Masters CL, Gajdusek DC (1980) Oral transmission of kuru, Creutzfeldt-Jakob disease, and scrapie to nonhuman primates. J Infect Dis 142: 205-208.
- Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218: 1309-1311.
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136-144.
- Kimberlin RH (1982) Scrapie agent: prions or virinos? Nature 297: 107-108.
- Manuelidis L, Yu ZX, Barquero N (2007) Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles. ProcNatlAcadSci 104: 1965-1970.
- Bandea C (2009) Endogenous Viral Etiology of Prion Diseases. Nature Precedings https://hdl.handle.net/10101/npre.2009.3887.1
- Miyazawa K, Emmerling K, Manuelidis L (2011) High CJD infectivity remains after prion protein is destroyed. J Cell Biochem 112: 3630-3637.
- Cohen FE, Prusiner SB (1998) Pathologic conformations of prion proteins. Annu Rev Biochem 67: 793-819.
- Colby DW, Prusiner SB (2011) Prions. Cold Spring HarbPerspectBiol 3: a006833.
- Maddelein ML1, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ (2002) Amyloid aggregates of the HET-s prion protein are infectious. ProcNatlAcadSci U S A 99: 7402-7407.
- Osherovich LZ, Weissman JS (2002) The utility of prions. Dev Cell 2: 143-151.
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, et al. (2007) The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J 26: 4038-4050.
- Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, et al. (2014) Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest 124: 847-858.
- Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, et al. (2013) Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. ProcNatlAcadSci U S A 110: 7044-7049.
- Nishizawa K, Oguma A, Kawata M (2014) Efficacy and Mechanism of a Glycoside Compound Inhibiting Abnormal Prion Protein Formation in Prion-Infected Cells: Implications of Interferon and Phosphodiesterase 4D-Interacting Protein J Virol 88: 4083-4099.
- Piccardo P, Cervenak J, Bu M (2014) Complex proteinopathy with accumulations of prion protein, hyperphosphorylated tau, a-synuclein and ubiquitin in experimental bovine spongiform encephalopathy of monkeys. J Gen Virol 95: 1612-1618.
- Mabbott NA (2015) Prospects for safe and effective vaccines against prion diseases. Expert Rev Vaccines 14: 1-4.
- Souan L, Tal Y, Felling Y, Cohen IR, Taraboulos A, et al. (2001) Modulation of proteinase-K resistant prion protein by prion peptide immunization. Eur J Immunol 31: 2338-2346.
- Kovalchuk Ben-Zaken O, Nissan I, Tzaban S, Taraboulos A, Zcharia E, et al. (2015) Transgenic over-expression of mammalian heparanase delays prion disease onset and progression. BiochemBiophys Res Commun 464: 698-704.
- Van Everbroeck B, Boons J, De Leenheir E, Lübke U, Cras P (2004) Molecular diagnostic tools in Creutzfeldt-Jakob disease and other prion disorders. Expert Rev MolDiagn 4: 351-359.
- Lehto MT, Peery HE, Cashman NR (2006) Current and future molecular diagnostics for prion diseases. Expert Rev MolDiagn 6: 597-611.
- McKinley MP, Bolton DC, Prusiner SB (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35: 57-62.
- Barry RA, Kent SB, McKinley MP, Meyer RK, DeArmond SJ, et al. (1986) Scrapie and cellular prion proteins share polypeptide epitopes. J Infect Dis 153: 848-854.
- McBride PA, Bruce ME, Fraser H (1988) Immunostaining of scrapie cerebral amyloid plaques with antisera raised to scrapie-associated fibrils (SAF). NeuropatholApplNeurobiol 14: 325-336.
- Safar J, Wille H, Itri V, Groth D, Serban H, et al. (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4: 1157-1165.
- Barnard G, Helmick B, Madden S, Gilbourne C, Patel R (2000) The measurement of prion protein in bovine brain tissue using differential extraction and DELFIA as a diagnostic test for BSE. Luminescence 15: 357-362.
- Grosset A, Moskowitz K, Nelsen C, Pan T, Davidson E, et al. (2005) Rapid presymptomatic detection of PrPSc via conformationally responsive palindromic PrP peptides. Peptides 26: 2193-2200.
- Chang B, Gray P, Piltch M, Bulgin MS, Sorensen-Melson S, et al. (2009) Surround optical fiber immunoassay (SOFIA): an ultra-sensitive assay for prion protein detection. J Virol Methods 159: 15-22.
- Bosque PJ, Prusiner SB (2000) Cultured cell sublines highly susceptible to prion infection. J Virol 74: 4377-4386.
- Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411: 810-813.
- Colby DW, Zhang Q, Wang S, Groth D, Legname G, et al. (2007) Prion detection by an amyloid seeding assay. ProcNatlAcadSci U S A 104: 20914-20919.
- Atarashi R, Sano K, Satoh K, Nishida N (2011) Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion 5: 150-153.
- Hadlow WJ, Eklund CM, Kennedy RC, Jackson TA, Whitford HW, et al. (1974) Course of experimental scrapie virus infection in the goat. J Infect Dis 129: 559-567.
- Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, et al. (1982) Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol 11: 353-358.
- Andréoletti O, Litaise C, Simmons H, Corbière F, Lugan S, et al. (2012) Highly efficient prion transmission by blood transfusion. PLoSPathog 8: e1002782.
- Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA (2009) Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One 4: e4848.
- Van Everbroeck B, Pals P, Martin JJ, Cras P (1999) Antigen retrieval in prion protein immunohistochemistry. J HistochemCytochem 47: 1465-1470.
- An SSA, Lim KT, Oh HJ (2010) Differentiating blood samples from scrapie infected and non-infected hamsters by detecting disease-associated prion proteins using multimer detection system. Biochemical and Biophysical Research Communications 392: 505-509.
- Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, et al. (2011) Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377: 487-493.
- Korth C, Stierli B, Streit P, Moser M, Schaller O, et al. (1997) Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 390: 74-77.
- Korth C, Stierli B, Streit P, Moser M, Schaller O, et al. (1997) Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 390: 74-77.
- Meyer RK, Oesch B, Fatzer R, Zurbriggen A, Vandevelde M (1999) Detection of bovine spongiform encephalopathy-specific PrP(Sc) by treatment with heat and guanidine thiocyanate. J Virol 73: 9386-9392.
- Lane A, Stanley CJ, Dealler S (2003) Polymeric ligands with specificity for aggregated prion proteins. ClinChem 49: 1774-1775.
- Serban D, Taraboulos A, DeArmond SJ, Prusiner SB (1990) Rapid detection of Creutzfeldt-Jakob disease and scrapie prion proteins. Neurology 40: 110-117.
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, et al. (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J MolBiol 273: 614-622.
- Bellon A, Seyfert-Brandt W, Lang W (2003) Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol 84: 1921-1925.
- Rubenstein R, Chang B, Gray P, Piltch M, Bulgin MS, et al. (2010) A novel method for preclinical detection of PrPSc in blood. J Gen Virol 91: 1883-1892.
- Laude H, Vilette D, Le Dur A, Archer F, Soulier S, et al. (2002) New in vivo and ex vivo models for the experimental study of sheep scrapie: development and perspectives. C R Biol 325: 49-57.
- Klöhn PC, Stoltze L, Flechsig E, Enari M, Weissmann C (2003) A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. ProcNatlAcadSci U S A 100: 11666-11671.
- Solassol J, Crozet C, Lehmann S (2003) Prion propagation in cultured cells. Br Med Bull 66: 87-97.
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, et al. (2001) Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. ProcNatlAcadSci U S A 98: 4055-4059.
- Castilla J, Saá P, Soto C (2005) Detection of prions in blood. Nat Med 11: 982-985.
- Thorne L, Terry LA (2008) In vitro amplification of PrPSc derived from the brain and blood of sheep infected with scrapie. J Gen Virol 89: 3177-3184.
- Moda F, Gambetti P, Notari S, Concha-Marambio L, Catania M, et al. (2014) Prions in the urine of patients with variant Creutzfeldt-Jakob disease. N Engl J Med 371: 530-539.
- Lacroux C, Comoy E, Moudjou M, Perret-Liaudet A, Lugan S, et al. (2014) Preclinical detection of variant CJD and BSE prions in blood. PLoSPathog 10: e1004202.
- Wadsworth JD, Joiner S, Hill AF (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358: 171-180.
- Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, et al. (2008) Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 5: 211-212.
- Orrú CD, Wilham JM, Raymond LD, Kuhn F, Schroeder B, et al. (2011) Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. MBio 2: e00078-00011.
- Orrú CD, Bongianni M, Tonoli G, Ferrari S, Hughson AG, et al. (2014) A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med 371: 519-529.

