Keywords
D2E7; Antibody engineering; Affinity; TNFα; Biological activity
Introduction
Monoclonal antibodies are widely used immunological tool with therapeutic, research, and also diagnostic applications because of high specificity and affinity against their antigens [1]. A variety of well-established techniques such as hybridoma, phage, and other associated display approaches, have been employed to develop numerous novel antibodies against potential targets [2,3]. Antibody engineering approaches are often necessary to optimize physicochemical characteristics of the bio-therapeutic agents regarding to clinical progress and industrial production. Various properties that potentially require to be improved are including specificity, affinity, stability, solubility, lack of aggregation, solubility, etc. [4]. Antibodies acquire their affinity and specificity towards a variety of target antigens by modifying amino acid residues within their six CDRs, three from heavy chain (CDR-H1, CDR-H2, CDR-H3) and three from light chain (CDR-L1, CDR-L2, CDR-L3) which known as hypervariable regions [5]. The molecules precisely recognize epitopes of varying size using only a limited number of amino acid residues in their CDRs [6]. Recently, complicates experimental and computational efforts have occurred to engineer the CDRs compositions and achieve therapeutic antibodies that recognize its antigen with higher affinity and specificity [7]. In most cases, an increase in the binding activity and affinity of antibody enhanced the biological activity and decreased the dose of requirements, lead to improved efficiency, lower side effects and costs.
Until now, various mutagenesis strategies have confirmed as useful methods to increase the affinity of antibodies by substitution of particularly selected residues in the CDR loops or by random mutagenesis in the variable fragment sequence [8,9]. Furthermore, several computational methods have been employed to improve affinity of different antibodies by using of rational insights into the CDR loops and reported that CDR-L3 and, especially, CDR-H3 are frequently responsible for greatest contacts between antibody-antigen [10-12].
TNFα is a multifunctional cytokine that play some crucial roles in regulating immune hematopoiesis and in developing inflammation diseases [13-15]. Accordingly, different anti-TNFα antibodies including etanercept, golimumab, infliximab, adalimumab and certolizumab, have been approved for treatment of several diseases such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis [16,17]. D2E7, a full-humanized antibody, developed by phage display method and is extensively used with rheumatoid arthritis patients. Former studies have focused on to reduce side effects of the antibody that is in result of highly recommended dose in the patients. In the studies, D2E7 have been applied as a target for CDR engineering in order to improved affinity and biological activity [8,18].
In our research group, we have applied a set of bioinformatics analyses based on molecular dynamics simulations approaches that reported with other study [19] to produce D2E7 mutant models with an improved binding affinity and biological activity (data not shown). According to our data, three variant of D2E7 antibody with a single point mutation at their CDRs of light chain (lc-A94K, lc-N92R, lc- A50Y), and a variant with a single point mutation at its CDR1 of heavy chain (hc-A33R) are potential models to be experimentally investigated in respect of increased affinity and potency.
In the present study, we aimed to evaluate binding affinity and biological activity of D2E7 antibody and the four candidate models against TNFα to find the best-optimized model with minimum structural modifications.
Methods
Expression vectors and cloning
DNA encoding the light and heavy chain of D2E7 antibody was codon optimized for antibody expression in mammalian cells. The expression vector pcDNA3.1 that containing D2E7 sequence was obtained from General Biosystem (USA). To prepare vectors expressing the mutant antibodies, we applied polymerase chain reaction (PCR) site-directed mutagenesis approach. Primers were used for the mutagenesis step are listed (Table 1). PCR was performed by using Pfu DNA polymerase with the following conditions: one cycle at 95°C for five minutes, followed by 18 cycles with 95°C for 50 seconds, 60°C for 50 seconds and 68°C for 9 minutes and at the end one cycle at 68°C for one minute. The PCR products were digested with 1 μl of Dpn1 enzyme (ThermoFisher, USA) and incubated for 10 minutes at 37°C. The mutated plasmids were transformed into competent cells of E. coli strain DH5α. The bacteria were grown overnight at 37°C on agar plates. Three single colonies were picked and expanded. Plasmids of each colonies were extracted using a Miniprep kit (ThermoFisher, USA) and sequenced (Microsynth, USA). To confirm mutagenesis, the purified plasmids were sequenced using 3130 ABI sequencer.
| Oligo Name |
Base Number |
Tm* (°C) |
Oligo Sequence |
| lc-A94K-P1 |
40 |
78 |
GCC TTG GCC GAA GGT GTA TGG TTT TCT GTT GTA TCT TTG G |
| lc-A94K-P2 |
40 |
78 |
CCA AAG ATA CAA CAG AAA ACC ATA CAC CTT CGG CCA AGG C |
| lc-N92R-P1 |
38 |
79 |
GGC CGA AGG TGT ATG GTG CTC TTC TGT ATC TTT GGC AG |
| lc-N92R-P2 |
38 |
79 |
CTG CCA AAG ATA CAG AAG AGC ACC ATA CAC CTT CGG CC |
| lc-A50Y-P1 |
35 |
79 |
CCG CTT TGC AGG GTA GAG GCG TAG TAG ATG AGG AG |
| lc-A50Y-P2 |
35 |
79 |
CTC CTC ATC TAC TAC GCC TCT ACC CTG CAA AGC GG |
| hc-A33R-P1 |
45 |
80 |
GCC TGT CTC ACC CAG TGC ATT CTA TAG TCG TCA AAT GTA AAT CCG |
| hc-A33R-P2 |
45 |
80 |
CGG ATT TAC ATT TGA CGA CTA TAG AAT GCA CTG GGT GAG ACA GGC |
*Tm: Primer Melting Temperature
Table 1: Primers of site directed mutagenesis.
Expression and purification of antibodies
Human embryonic kidney cells 293 (HEK293T) (ATCC, LGC Standards GmbH, Wesel, Germany: ATCC-No. CRL-11268) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermofisher, USA) supplemented with 8% (v/v) heat inactivated fetal calf serum (FCS) (Gibco Sera, Thermo Fischer, USA) and 1% (v/v) of a 5.000 U/mL penicillin-streptomycin solution (Thermfisher, USA) at 37°C, 5% CO2 and 95% humidity [20]. For transfection, HEK293T cells were seeded into 10 cm plates using 12.5 mL culture medium so that they reached 70-80% confluence after one day in culture. In the next step, 10 μg polyethyleneimine (PEI) (Polysciences, USA), and 10 μg high quality purified plasmid were separately diluted in 600 μL DMEM medium. The DNA and PEI solutions were combined, mixed and incubated for 15–30 minutes at room temperature (RT). The DNA/PEI mixtures were distributed over the HEK293T cells and incubated for 24 h at 37°C. Following day, the medium was completely replaced with DMEM medium that was supplemented with 1% penicillin/streptomycin and with 4% (v/v) of IgG stripped FCS (PAA) to minimize co-purification of bovine IgG. The medium was exchanged and harvested every day for up to one week and the expression of the antibody was monitored using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12% polyacrylamide gel.
Protein-A affinity chromatography
Protein-A affinity purification was performed using 1 ml HiTrap MabSelect SuRe columns (GE Healthcare, USA) on an ÄKTA protein purification system (GE Healthcare, USA). This column is specified for the purification of monoclonal antibody based on their affinity for protein-A. The binding and wash buffers were as follows: 20mM sodium phosphate, 0.15 M NaCl (pH: 7.2). Elution buffer was 0.1M sodium citrate (pH: 3.0) and neutralization buffer was 1M Tris-HCl (pH: 9). Concentration of the antibodies was determined by optical density at 280 nm on an ultraviolet (UV)–light spectrophotometer (GE Healthcare, USA) using the extinction coefficient (EC) of the antibody. The purified antibodies were analyzed by SDS-PAGE on a 12% polyacrylamide gel under reducing and non-reducing conditions.
Circular Dichroism (CD) measurement
CD spectroscopy was carried out on a JASCO J-810 spectrometer to compare the secondary structure of mutant and wild antibodies, at 0.125 mg/ml, in the far-UV region (250–190 nm), at 25°C. Three consecutive scans were accumulated for each sample and the spectra obtained were averaged. The Mean residue ellipticity (MRE) was calculated according to the previous works [21] and the BESTSEL server [22].
Enzyme-Linked Immunosorbent Assay (ELISA) measurement
Binding activities of the native and mutant antibodies to recombinant human TNFα (Biolegend, USA) were determined by ELISA. First, 100 μl/well TNFα (1 μg/ml) in phosphate buffered saline (PBS, pH=7.2) was coated at a 96-well microtitre plate and incubated overnight at 4°C. After that, the wells were washed three times with PBS containing 0.05% Tween-20 (PBST) and blocked with blocking buffer (PBS containing 1% FCS) for 2 hr at RT. Following, the antibodies (diluted in PBS) were added to the wells in serial dilution, triplicates and then incubated for 2 hr at RT. The wells were washed four times with PBST. A total of 100 μl of horseradish peroxidase-conjugated Goat- Anti-Human IgG (Abcam, USA) was added to each well and incubated for 1 hr at RT. After five washing steps by PBST, 100 μl of substrate 3,3´,5,5´,-tetramethylbenzidine (TMB) (Biomol, Germany) was added as a substrate to the wells and the absorbance was read at 450 nm on a microplate reader.
Surface Plasmon Resonance (SPR) measurement
The kinetic affinity of the native and mutant antibodies to recombinant human TNFα was determined by SPR spectroscopy on a Biacore X100 (GE Healthcare, USA) instrument. The assay format was Fc-based capture via immobilized antibody. A ready to use carboxymethylated dextran (CM5) sensor chip was built up by a carboxymethylated dextran matrix with a recombinant protein-A variant that covalently attached to it. The chip eliminated needs for developing an immobilization procedure. In a first step, antibody (5 μg/ mL) was bound to this protein-A sensor chip (GE Healthcare, USA) with a flow rate of 10 μL/minute for 180 seconds to reach an optimum response unit (RU). Afterward, different concentrations of TNFα (200, 100, 50, 25, 12.5, 6.25, 3.125, 1.56 and 0.00 nM) were injected at a flow rate of 80 μL/minute for 180 seconds (240 μL of analyte) to allow their association with the bound antibodies. For the dissociation of the TNFα/ antibody complexes, running buffer contain HBS-EP buffer pH 7.4 [0.01 M HEPES (4-{2-hydroxyethyl-1-piperazineethanesulfonic acid), 0.15 M NaCl, 3 mM Ethylenediaminetetraacetic acid (EDTA) and 0.005% v/v Surfactant P20] (GE Healthcare, USA) was injected at a flow rate of 80 μL/minute for 15 minutes. Biosensor matrices were regenerated using regeneration buffer (glycine-HCl, pH 1.5) (GE Healthcare, USA). Data were processed and fitted to a 1:1 binding model using Biacore X100 evaluation software (version 1.0) to determine the binding kinetic rate constants, ka (on-rate) and kd (off-rate), and KD (equilibrium dissociation constant). We evaluated the likelihood of fitting by the Chi2 and U-value statistics. The significance threshold of the parameters for U-value is<25 as reported elsewhere [23].
TNFα neutralizing activity
TNFα neutralizing activity of the antibodies evaluated on murine fibroblast L929 cells (IBG1, KIT, Germany). The cells were grown in DMEM medium supplemented with 10% FCS and then seeded at 200,000 cells/well in 96-well plate overnight at 37°C, triplicate. A series of diluted antibody samples were prepared in the presence of a fixed concentration of TNFα (10 ng/ml) and actinomycin D (Sigma, USA) for 60 minutes at 37°C in order to allow neutralization effect of the antibodies would occur. After the neutralization, 100 μL of antibody/ TNFα mixture was added to each well. The cell plates were further incubated at 37°C for 24 hours. After that, TNFα-neutralizing activity was estimated by a 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) (M200, SIGMA) assay. The medium in each well was replaced with 200 μl MTT solution (5 μg/ml) and the cell plates were incubated for 4 hours at 37°C. Then, the supernatant was removed and tetrazolium salts that formed into the living cells were solubilized with 100 μl of dimethyl sulphoxide (DMSO) (Sigma-Aldrich). They were shaken for 10 minutes and the absorbance of each well was measured at 570 nm by employing a microplate reader (Biotek, ELx 800, and USA). Results achieved as a percent of cell viability were plotted against different concentrations (ng/mL) of samples applied by using GraphPad Prism 7 software.
Structural analysis of binding interactions
The X-ray crystallographic structure of D2E7/TNFα complex (3WD5.pdb) was found from Protein Data Bank [24]. The 3WD5.pdb was used as an input to Pymol software [25], then the structure was mutated by mutagenesis option of Pymol to generating 3D structure of lc-A94K antibody and then was energy minimized by Gromacs version 4.5.6 [26]. LigPlot was used to evaluating binding pocket of the antibody-antigen complexes [27]. Ligplot is a program to produce graphic illustrations of protein-ligand interactions including hydrogen-bond and hydrophobic interaction between the ligand(s) and the main-chain or side-chain elements of the protein [28].
Statistical analysis
Obtained data were statistically analyzed with GraphPad Prism 7 software. For in vitro assays, the statistical analyses performed using two-way analysis of variance (ANOVA). For all statistical analyses, a value of p<0.05 was regarded as statistically significant.
Results
Cloning and expression of the antibodies
The pcDNA3.1 vector with the whole sequence of the D2E7 antibody was used to produce D2E7 antibody. For the mutant antibodies, we introduced mutations using the four paired primers (Table 1) and site directed mutagenesis method [29]. Plasmids encoding the antibodies were transfected into HEK293T cells using PEI. The expression of the antibodies was confirmed by SDS-PAGE. We used Cinorra (a commercial form of D2E7) as a positive control. In non-reducing SDS-PAGE, we observed a 170 Kilodalton (kDa) protein band for all the antibodies (Figure 1A). In reducing SDS-PAGE, two bands at 55 kDa and 27 kDa corresponding to the light and heavy chains of the antibodies appeared in all samples (Figure 1B), which proved that our expressed antibodies behaved exactly the same way as the commercial Cinorra antibody do. We harvested the cell culture supernatant of day 1,2 and 3 and purified them by using protein-A affinity chromatography. Purity of the samples was confirmed by SDS-PAGE under both non-reducing and reducing condition (Figure 1C and 1D).
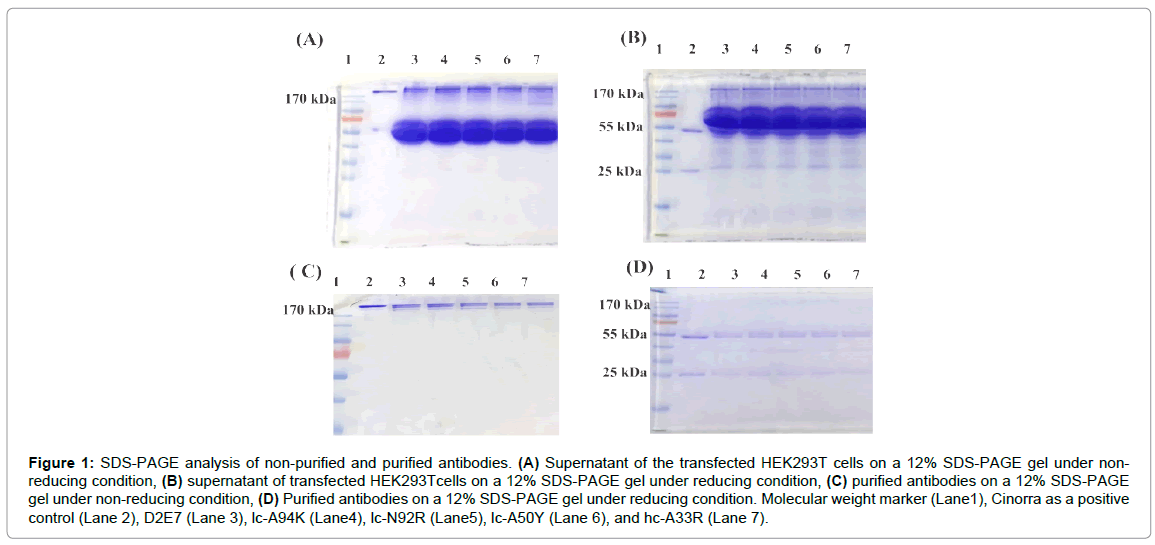
Figure 1: SDS-PAGE analysis of non-purified and purified antibodies. (A) Supernatant of the transfected HEK293T cells on a 12% SDS-PAGE gel under nonreducing condition, (B) supernatant of transfected HEK293Tcells on a 12% SDS-PAGE gel under reducing condition, (C) purified antibodies on a 12% SDS-PAGE gel under non-reducing condition, (D) Purified antibodies on a 12% SDS-PAGE gel under reducing condition. Molecular weight marker (Lane1), Cinorra as a positive control (Lane 2), D2E7 (Lane 3), lc-A94K (Lane4), lc-N92R (Lane5), lc-A50Y (Lane 6), and hc-A33R (Lane 7).
Analysis of the secondary structures
To gain further information about possible conformational effects of the mutations, secondary structure patterns of the antibodies were evaluated by CD spectroscopy in the far-UV range (190-260 nm). The CD spectra of the wild and the mutant antibodies possessed an identical line shape and reveal a dominance of β-sheet structures (Figure 2). Consequently, the data suggest that the mutations have no remarkable impact (p>0.05) on the secondary structure of D2E7 (Table 2).
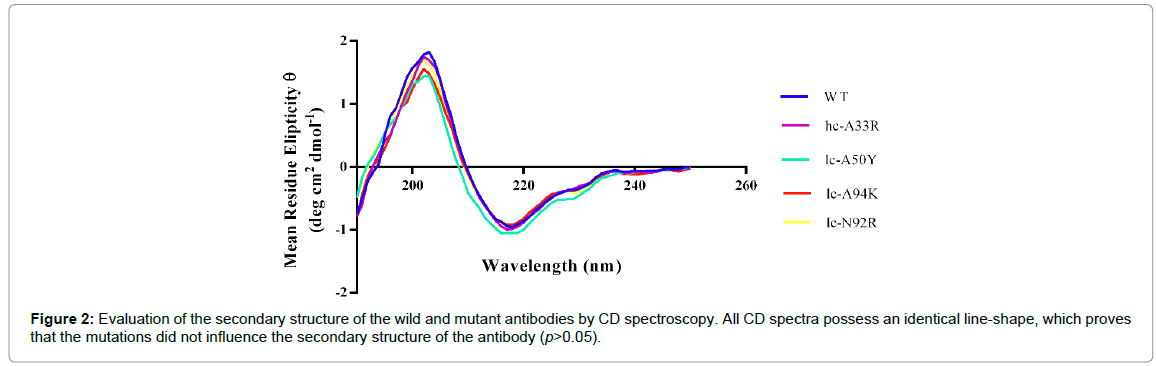
Figure 2: Evaluation of the secondary structure of the wild and mutant antibodies by CD spectroscopy. All CD spectra possess an identical line-shape, which proves that the mutations did not influence the secondary structure of the antibody (p>0.05).
| Antibodies |
Helix |
Antiparallel |
Parallel |
Turn |
Others |
Anti 1 (Left twisted) |
Anti 2 (Relaxed) |
Anti (Right-twisted) |
| Wild |
0 |
46 |
7.7 |
10.3 |
36 |
7.6 |
19 |
19.4 |
| lc-A94K |
0 |
44.9 |
7.9 |
10.9 |
36.3 |
7.1 |
18.5 |
19.3 |
| hc-A33R |
0 |
46.1 |
7.8 |
10.6 |
35.5 |
7.5 |
19.3 |
19.3 |
| lc-A50Y |
0.5 |
44.7 |
8.1 |
11 |
35.7 |
7.3 |
17.8 |
19.6 |
| lc-N92R |
0 |
46.4 |
6.4 |
11.1 |
36.1 |
7.5 |
19.1 |
19.8 |
Table 2: Quantitative analysis of the secondary structure of the wild and mutant antibodies by CD spectroscopy.
Evaluation of the binding activity of the antibodies
ELISA assay was performed to evaluate ability of the wild and the mutant antibodies in binding to recombinant human TNFα by determining the concentration value at which the antibodies neutralize 50% of the antigens (EC50). As demonstrated in Figure 3, the EC50 of lc-A94K (194.1 ng/ml) and lc-N92R (240.7 ng/ml) increased as 215 and 173 percent, respectively, in comparison to the EC50 of the wild antibody (416.5 ng/ml). The EC50 of lc-A50Y (774.6 ng/ml) and hc- A33R (917.4 ng/ml) increased in comparison to the native antibody, which indicates a lower binding activity of these mutants to TNFα. As such, the EC50 values revealed that lc-A94K and lc-N92R models significantly improved their binding potency for interaction to TNFα (p<0.003) (Figure 3). We candidate these two models for SPR analysis and biological assay.
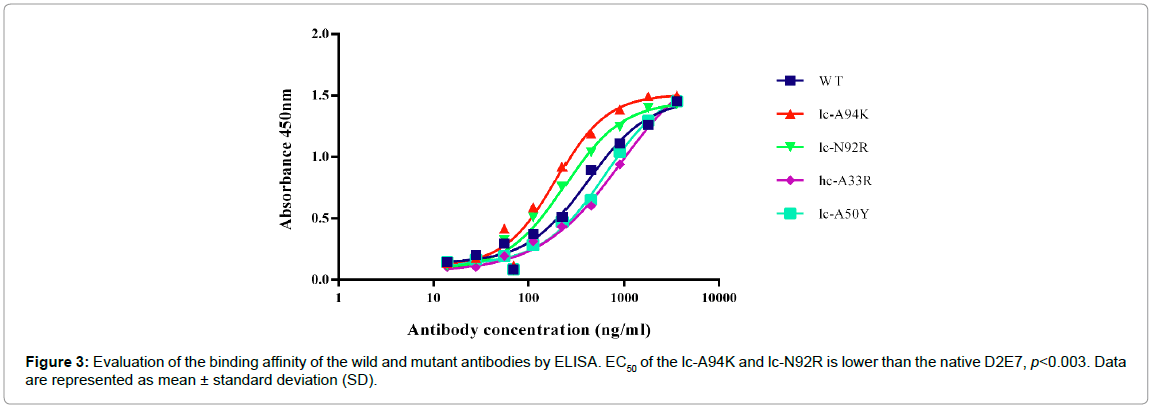
Figure 3: Evaluation of the binding affinity of the wild and mutant antibodies by ELISA. EC50 of the lc-A94K and lc-N92R is lower than the native D2E7, p<0.003. Data are represented as mean ± standard deviation (SD).
Evaluation of the kinetics affinity of the antibodies
To get further insight into kinetic affinity, SPR spectroscopy was performed to measure the thermodynamic affinity and the kinetic constant of lc-A94K and lc-N92R antibodies in comparison to the native D2E7. In the first step, a fixed concentration of antibodies was immobilized on a protein-A sensor chip [30]. The following injection of TNFα to this pre-coated surface generated a RU of the SPR signal, which is related to the Ka. The Kd was obtained by the dissociation of the antigen from the antibody surface during washing, which leads to a decrease of the RU signal. Binding constant or KD is the average of three experiments. The native antibody KD was as 0.38 nM (Table 3). Further, it was evident that the A94K mutation caused a 240% increase in kinetics affinity of the mutant model compared to the native monoclonal antibody (KD=0.16 nM) (Figure 4A and 4B). The increase in affinity of lc-A94K for TNFα is due to enhanced on-rates compared with native D2E7 antibody (Table 3). This further indicated a significant improvement (p<0.005) of the binding affinity of the mutant antibody for TNFα. Furthermore, the KD of the lc-N92R mutant (KD=0.37 nM) is only a bit different in comparison to wild antibody, which means that the mutation had no effect in terms of improving the antigen binding.
| Antibody |
Ka (M-1S-1) |
Kd (S-1) |
*KD (M) |
?U-value |
| Wild |
3.6 E+5 (1.1E+05) |
1.4 E-4 (3.2E-7) |
3.8E-10 (2.2E-11) |
12 |
| lc-A94K |
6.8 E+5 (1.0 E+05) |
1.1 E-4 (2.1E-7) |
1.6E-10 (1.4E-11) |
3 |
| lc-N92R |
4.9 E+5 (1.3 E+05) |
1.8 E-4 (4.2E-7) |
3.7E-10 (1.7E-11) |
12 |
*Binding constants (KD) are the average of three experiments (SD in parenthesis).
?U-value significant threshold is<25.
Table 3: Affinity and binding kinetic parameters of wild and mutant antibodies.
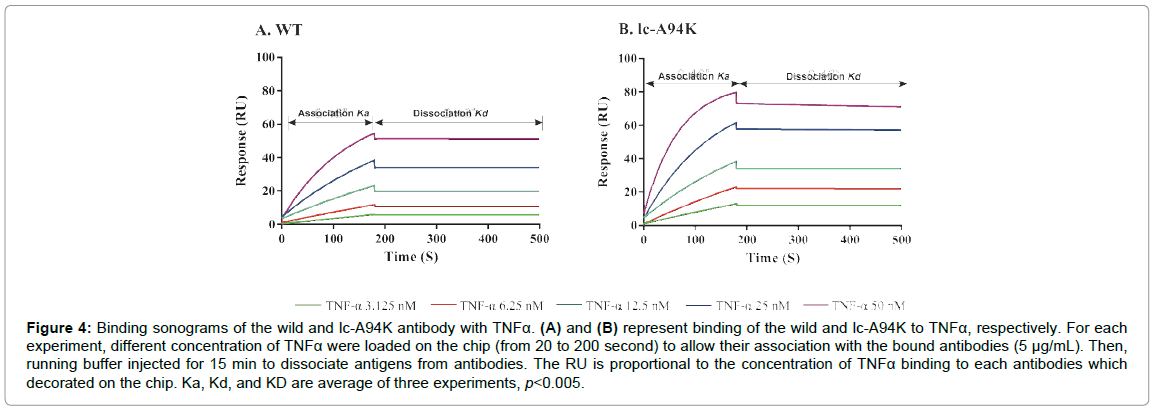
Figure 4: Binding sonograms of the wild and lc-A94K antibody with TNFa. (A) and (B) represent binding of the wild and lc-A94K to TNFa, respectively. For each experiment, different concentration of TNFa were loaded on the chip (from 20 to 200 second) to allow their association with the bound antibodies (5 µg/mL). Then, running buffer injected for 15 min to dissociate antigens from antibodies. The RU is proportional to the concentration of TNFa binding to each antibodies which decorated on the chip. Ka, Kd, and KD are average of three experiments, p<0.005.
Neutralization of TNFα-mediated cytotoxicity by the antibodies
We evaluated biological activity of the lc-A94K, lc-N92R and wild antibodies to neutralized cytotoxicity effect of TNFα in L929 cells with MTT colorimetric assay. The viability of L929 cells that were pre-treated with TNFα, analyzed by co-incubation of the cells with each antibody in separate experiments. We found lc-A94K and native D2E7 antibody could efficiently neutralize cytotoxicity in L929 cells through a dose-dependent manner, with IC50 of 450 ng/ml and 145 ng/ml, respectively. As shown in Figure 5, lc-A94K antibody neutralizes cytotoxicity of the cells significantly three fold stronger than the native D2E7 (p<0.001). These results, taken together, confirmed that single point mutation of ALA to LYS in the lc-CDR3 D2E7 increase binding activity of the antibody for TNFα results in improvement of its potency to neutralize cytotoxicity effect of TNFα. The IC50 of lc-N92R was about 470 ng/ml that means the biological activity of the mutant decreased in contrast to wild antibody (Figure 5).
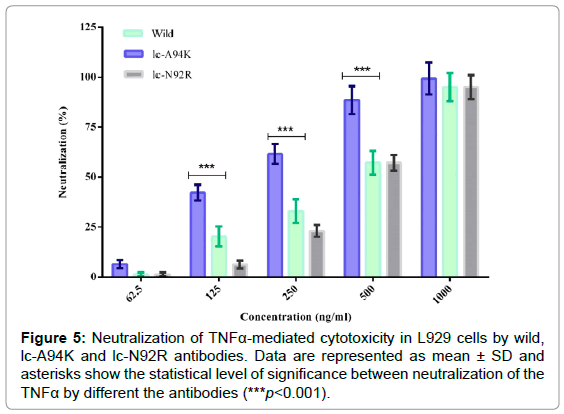
Figure 5: Neutralization of TNFa-mediated cytotoxicity in L929 cells by wild, lc-A94K and lc-N92R antibodies. Data are represented as mean ± SD and asterisks show the statistical level of significance between neutralization of the TNFa by different the antibodies (***p<0.001).
Binding interaction analysis of antibodies in complex with TNFα
The 3D structure of D2E7/TNFα complex and lc-A94K/TNFα complex were applied as an input for Ligplot software. The program visualized hydrogen and hydrophobic interactions at the binding pocket of the complexes. By analyzing the antibody-antigen complexes, we found that mutating of ALA to LYS in the lc-CDR3 of D2E7 antibody leaded to generating new hydrogen bonds and hydrophobic interactions with the epitopes of TNFα. The data showed ARG90 and ARG93 from wild antibody are participating in the three hydrogen bonds with PRO20, GLN21 and ALA22 from TNFα (Figure 6A). Remarkably, in the binding pocket of lc-A94K/TNFα complex, ARG90, ARG93 and ARG94 from lc-A94K are taking part in the six hydrogen bonds with GLN21, ALA22, PRO20, GLU23, and ALA145 from TNFα (Figure 6B).
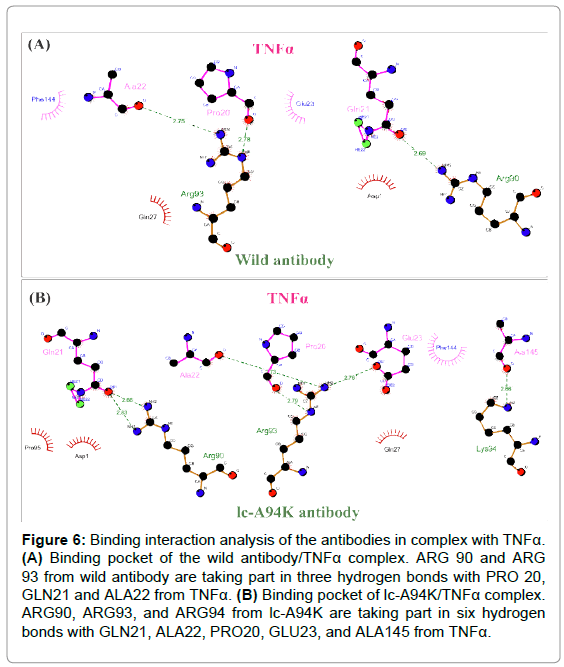
Figure 6: Binding interaction analysis of the antibodies in complex with TNFa. (A) Binding pocket of the wild antibody/TNFa complex. ARG 90 and ARG 93 from wild antibody are taking part in three hydrogen bonds with PRO 20, GLN21 and ALA22 from TNFa. (B) Binding pocket of lc-A94K/TNFa complex. ARG90, ARG93, and ARG94 from lc-A94K are taking part in six hydrogen bonds with GLN21, ALA22, PRO20, GLU23, and ALA145 from TNFa.
Discussion
Adalimumab is a human recombinant IgG1 isotype directed against the soluble and cell-bound forms of TNFα [31]. Although the antibody has already high affinity but patients require a high dosage of the antibody per single administration that such dosage can intensify different side effects [32-34]. Various studies revealed greater binding affinity, particularly for soluble cytokines, often correlates with improved biological activity, resulting in decrease dosage and potentially fewer adverse events [35,36]. Thus CDRs engineering of the antibody is a promising starting point for increasing its binding affinity to TNFα regarding to achieve greater biological activity and clinical efficiency. With similar reasoning, we have applied a computational structural approach to making D2E7 mutants with improved binding affinity to TNFα. In the current study, we cloned and expressed lc- A94K, lc-N92R, lc-A50Y, hc-A33R mutant models and D2E7 antibody in HEK293T cell line. We identified that the EC50 and KD of the lc- A94K mutant were significantly increased in comparison to the EC50 and KD of the native antibody. Moreover, our data revealed that lc- A94K antibody could effectively neutralize the cytotoxicity effect of TNFα in L929 cells with higher potency than the native antibody.
In the present study, we aimed to improve the binding affinity of D2E7 to its ligand by introducing a minimum number of mutations (single mutations) in CDR sequences while preserving the native structure of the antibody as much as possible. We found that the secondary structures of all mutants are consistent with the structures of wild antibody (Table 2). These data showed the mutations occurred without affecting the secondary structures of the mutant antibodies thus the principle structure of D2E7 have been precisely conserved at the mutant models. Rajpal et al. engineered D2E7 CDRs by introducing more than ten point mutations in order to increase binding affinity [8]. In another similar study, Votsmeie et al. used a display-based approach in order to enhance the affinity of D2E7 antibody [18]. Although the two groups had been successful to significantly improve the D2E7 affinity, recent studies revealed that primary problem by introducing extensive mutations during affinity maturation is the modification of antibody basic structure that would translate to the adverse effects on their specificity and stability [37].
It is also notable that lc-A94K revealed an enhanced binding activity and kinetic affinity nearly 215 and 240 percent respectively in comparison to the wild antibody. Moreover, biological activity of the antibody improved three fold higher than the native antibody.
Interestingly, the mutant models with the greatest efficiency harbored a single point mutation that was modified with a charged amino acid residue. It was reported that the affinity of an antibody against its ligand was enhanced by introducing charged amino acid residues into the CDRs of an antibody [38]. From a structural point of view on the binding site of lc-A94k with its antigen, we found mutation of ALA to LYS in residue94 of lc-CDR3 D2E7 enhanced hydrogen and hydrophobic interactions at the binding site of the antibody. Thus improvement of the non-polar bonds could potentially enhance energetically interactions at the binding pocket of lc-A94K/TNFα. Peng et al. revealed that energetically critical epitope factors have mainly consisted of hydrogen and hydrophobic bonds donors/acceptors. The physicochemical features are mainly important portions for paratopes of an antibody to recognizing an antigens [39]. Erijman et al. found that hydrogen (H) interactions are an important contributor to protein stability, folding and binding. They reported that although the energy of one H bond is relatively small, a large number of H bonds in proteins help them to play a significant role in protein-protein interaction energy [40].
In conclusion, we were enabled to identify lc-A94K as a mutant model of D2E7 antibody that contributed to an increase in binding affinity and biological activity of the antibody toward TNFα. Importantly, it should be pointed out that further in vivo and clinical assays are needed to compare therapeutic efficacy of lc-A94K with the original form of the antibody. The other mutant models showed no improvement in binding and potency to the ligand in the experimental assays. This may indicate that the affinity maturation is still a multipart complex process.
Acknowledgments
The authors wish to express their deep gratitude to Prof. Dr. Anne S. Ulrich, Prof. Dr. Christof Niemeyer, Dr. Jochen Bruck, Dr. Ruben Garrecht, Dr. Davami, Dr. Behrouz Vaziri, Timo Strunk, Dr. Sergiy Afonin, Christina Bauer, Dr. Tim Scharnweber and Seham Elabd who provided insight, expertise and technical support that greatly assisted the research.
Funding Source
This research was financially supported by a Pasteur Institute of Iran grant (for PhD Thesis of Maryam Tabasinezhad, Grant number: BP_9146) and Karlsruhe Institute of Technology.
24428
References
- Hudson P, Souriau C (2003) Engineered antibodies. Nature Med 9: 129-134.
- Bradbury A, Sidhu S, Dübel S, McCafferty J (2011) Beyond natural antibodies: The power of in vitro display technologies. Nature Biotech 29: 245-254.
- Miersch S, Sidhu S (2012) Synthetic antibodies: Concepts, potential and practical considerations. Methods 57: 486-498.
- Chiu M, Gilliland G (2016) Engineering antibody therapeutics. Curr Opin Struct Biol 38: 163-173.
- Kabat E, Wu T, Bilofsky H (1976) Some correlations between specificity and sequence of the first complementarity-determining segments of human kappa light chains. Proc Natl Acad Sci 73: 4471-4473.
- Kabat E, Wu T, Bilofsky H (1977) Unusual distributions of amino acids in complementarity determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J Biol Chem 252: 6609-6616.
- Rathore A, Sarker A, Gupta R (2018) Recent developments toward antibody engineering and affinity maturation. Protein Pept Lett 25: 886-896.
- Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, et al. (2005) A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc Natl Acad Sci 102: 8466-8471.
- Presta L (2006) Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Del Rev 58: 640-656.
- Arkadash V, Yosef G, Shirian J, Cohen I, Horev Y, et al. (2017) Development of high affinity and high specificity inhibitors of matrix metalloproteinase 14 through computational design and directed evolution. J Biol Chem 292: 3481-3495.
- Farhadi T, Fakharian A, Hashemian S (2017) Affinity improvement of a humanized antiviral antibody by structure-based computational design. Int J Pept Res Ther 25: 181-186.
- Grisewood M, Hernández-Lozada N, Thoden J, Gifford N, Mendez-Perez D, et al. (2017) Computational redesign of acyl-ACP thioesterase with improved selectivity toward medium-chain-length fatty acids. ACS Catalysis 7: 3837-3849.
- Tilg H (1997) New insights into the mechanisms of interferon alfa: an immunoregulatory and anti- inflammatory cytokine. Gastroenterology 112: 1017-1021.
- Baugh J, Bucala R (2001) Mechanisms for modulating TNF alpha in immune and inflammatory disease. Curr Opin Drug Discover Devel 4: 635-650.
- Bradley J (2008) TNF‐mediated inflammatory disease. J Pathol 214: 149-160.
- Wang Y, Wang H, Jiang J, Zhao D, Liu Y (2016) Comparative efficacy and acceptability of anti-TNF-alpha therapy in ankylosing spondylitis: a mixed-treatments comparison. Cell Physiol Biochem 39: 1679-1694.
- Stevenson M, Archer R, Tosh J, Simpson E, Everson-Hock E, et al. (2016) Adalimumab, etanercept infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: Systematic review and economic evaluation. Health Technol Assess 20: 1-610.
- Votsmeier C, Plittersdorf H, Hesse O, Scheidig A, Strerath M, et al. (2012) Femtomolar Fab binding affinities to a protein target by alternative CDR residue co-optimization strategies without phage or cell surface display. Mabs 4: 341-348.
- Kiyoshi M, Caaveiro J, Miura E, Nagatoishi S, Nakakido M, et al. (2014) Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS ONE 9: e87099.
- Jäger V, Büssow K, Wagner A, Weber S, Hust M, et al. (2013) High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol 13: 52.
- Kelly S, Jess T, Price N (2005) How to study proteins by circular dichroism. Biochim Biophysica Acta 1751: 119-139.
- Micsonai A, Wien F, Kernya L, Lee Y, Goto Y, et al. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci 112: 3095-3103.
- Canziani G, Klakamp S, Myszka D (2004) Kinetic screening of antibodies from crude hybridoma samples using Biacore. Anal Biochem 325: 301-307.
- Hu S, Liang S, Guo H, Zhang D, Li H, et al. (2013) Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor α-associated diseases from a molecular view. J Biol Chem 288: 27059-27067.
- DeLano W (2002) The PyMOL Molecular Graphics System. Delano Scientific.
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark A, et al. (2005) GROMACS: Fast, flexible, and free. J Comput Chem 26: 1701-1718.
- Wallace A, Laskowski R, Thornton J (1995) LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot Eng Des Select 8: 127-134.
- Laskowski R, Swindells M (2011) LigPlot+: Multiple ligand–protein interaction diagrams for dru Discovery. J Chem Inf Model 51: 2778-2786.
- Picard V, Ersdal-Badju E, Lu A, Bock S (1994) A rapid and efficient one-tube PCR-based mutagenesis technique using Pfu DNA polymerase. Nucleic Acids Res 22: 2587-2591.
- Bandyopadhyay S, Mahajan M, Mehta T, Singh A, Parikh A, et al. (2015) Physicochemical and functional characterization of a biosimilar adalimumab ZRC-3197. Biosimilars 5: 1-18.
- Rau R (2002) Adalimumab (a fully human anti-tumour necrosis factor α monoclonal antibody) in the treatment of active rheumatoid arthritis: The initial results of five trials. Ann Rheum Dis 61: ii70-ii73.
- Murdaca G, Colombo B, Puppo F (2011) Adalimumab for the treatment of immune-mediated diseases: An update on old and recent indications. Drug Today 47: 277-288.
- Tsilimbaris M, Diakonis V, Naoumidi I, Charisis S, Kritikos I, et al. (2009) Evaluation of potential retinal toxicity of adalimumab (Humira). Graefe's Arch Clin Exp Ophthalmol 247: 1119-1125.
- Scheinfeld N (2005) Adalimumab: A review of side effects. Expert Opin Drug Saf 4: 637-641.
- Zhou Y, Goenaga A, Harms B, Zou H, Lou J, et al. (2012) Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Mol Cancer Ther 11: 1467-1476.
- Carter P (2006) Potent antibody therapeutics by design. Nature Rev Immunol 6: 343-357.
- Tiller K, Chowdhury R, Ludwig S, Sen S, Maranas C, et al. (2017) Facile affinity maturation of antibody variable domains using natural diversity mutagenesis. Front Immunol 8: 986.
- Fukunaga A, Tsumoto K (2013) Improving the affinity of an antibody for its antigen via long-range electrostatic interactions. Protein Eng Des Sel 26: 773-780.
- Peng H, Lee K, Jian J, Yang A (2014) Origins of specificity and affinity in antibody–protein interactions. Proc Natl Acad Sci 111: 2656-2665.
- Erijman A, Rosenthal E, Shifman J (2014) How structure defines affinity in protein-protein interactions. PLoS ONE 9: e110085.


