Keywords
Myasthenia; Myasthenia crisis; AchR-Ab; Neuromuscular junction disorder; Anti-MuSK-Ab
Introduction
Myasthenia gravis (MG) is an autoimmune disorder with an average prevalence of 15-20 per 100,000 in the US population [1]. US Census Bureau, International Data Base 2004 estimated Myasthenia prevalence in United Arab Emirates to be 5 per 100,000. MG is purely an antibody mediated autoimmune disorder depicted by presence of auto-antibodies directed against post synaptic membrane of neuromuscular junctions. This is a T-cell mediated immune phenomenon occurring due to loss of tolerance to self-antigens [2]. Diagnosis of the disease is made on the basis of suggestive history of fluctuating fatigability especially if involving oculo-bulbar muscles, relevant clinical examination and a positive test. Tests include electrophysiological testing, pharmacological testing with edrophonium and/or laboratory detection of auto antibodies [3]. The most frequently tested antibodies are antiacetylcholine receptor (AChR) and anti-muscle-specific tyrosine kinase (MuSK) antibodies [4]. Disease mostly involves ocular muscles, oropharyngeal muscles or take a more generalized form [5].
About one-fifth of patients with MG experience crisis, usually within the first year of their illness. This is acute worsening of the disease. Respiratory failure is a potentially life-threatening complication that occurs in approximately 15 to 20% of patients diagnosed with MG [5]. It results from weakness of upper airway muscles leading to obstruction and aspiration or weakness of respiratory muscles leading to reduced tidal volumes, or from weakness of both. Over the past forty years, mortality rate has improved in myasthenia crisis from 75% to less than 5% [6].
Disease severity of MG can be assessed objectively using Osserman’s classification of Myasthenia gravis or Myasthenia Gravis Foundation (MGFA) of America grading [7,8]. In both classifications, disease is classified on the basis of group of muscles involved or progression and severity of disease.
Objectives of the study were to ascertain demographic details of myasthenia patients, to collect the clinical features especially myasthenic crisis (in terms of Myasthenia gravis foundation of America (MGFA) grading) and to correlate these with variety of antibodies.
Materials and Methods
A cross sectional study, based on record review of patients was carried out in Rashid Hospital, Dubai. Data was collected after a formal approval from Dubai Health Authority ethical committee. All the patients diagnosed with myasthenia gravis, visiting hospital were thoroughly recruited from 2004-2010 with at least five years of follow up.
Diagnosis of myasthenia was based on:
• History and clinical examination suggesting a fluctuating fatigability due to muscle tiredness and improvement of symptoms after rest.
• 15% response decrease in stimulation potential after repeated nerve stimulation.
• Improvement of muscle contraction edrophonium stimulation test.
• Single fiber electromyography.
Data was reviewed in detail by using records and hospital incorporate computer system. Results pertaining to qualitative variables were interpreted in the form of frequency and percentages. Distribution of sample was skewed with more incidence in early age group as well as non-uniform distribution among Osserman’s classification sub-groups. Thus, chi-square test was applied to ascertain association of autoimmune profile, clinico-therapeutic aspects and comorbidities. Data was analyzed using SPSS Version 21.
Results
A total of 143 patients, who visited our tertiary care hospital from 2004 to 2010 and then a follow up of minimum 5 years was carried out. Females predominated (61.8%) with a male to female ratio of 1:1.6. Age of our patients ranged from 15-68 years (mean = 34.5 ± 4 years). Majority of them had onset at younger age. As Dubai is the multicultural state, our patients belonged to various ethnicities comprising of UAE (57.8%), Arab countries other than UAE (22.5%), Asian (16%), Caucasian (3%) and African (1%) but for the statistical calculations we dichotomized them into Arabs (80.2%) and Non-Arab (19.8%).
Major part of UAE population consists of expat residents, so over the 5 years 41 patients lost follow up, probably they left for their home countries and we thoroughly followed 102 MG patients.
MG onset in 67.6% patients occurred at early age (before 35years), in female it was pretty earlier (<35 years of age), whereas in males it started mainly in late adult or elderly age. (p=0.001)
Clinically 16.7% patients had ocular and 83.3% had general form of myasthenia. Females predominantly had general myasthenia (88%). Ocular myasthenia had relatively earlier onset. As MG symptomatology bears wide range of variability and severity so it was graded according to Osserman’s classification and Myasthenia gravis foundation of America’s (MGFA) classification. Out of our patients 13.7% belonged to Osserman’s class-1, 43% to class-2, 37.3% to class-3, 2.9% to class-4 and 2.9% patients were categorized as class-5. Higher the Osserman’s class at presentation severer were the myasthenia crisis (p=0.006) but as number of observed patients at few places were less than 5 so statistical validity of chi-square test is not absolute. Account of clinical features delineated presence of motor symptoms in 75.5%, bulbar complaints in 54.9%, ocular symptoms in 82.4% and respiratory difficulties were in 14.7% patients. Immunologically motor and ocular features did not show exclusive specificity towards any particular antibodies, however bulbar features predominated in seropositive MG 63% vs. 25% (p=0.016).
As our prime target was to ascertain various types of autoantibodies and determine their role in clinical manifestation, auto-antibody evaluation showed Acetylecholine receptor antibody (AchR-Ab) in 75.5%, Muscle specific kinase antibodies (MuSK-Ab) in 7% patients and 17.6% did not contain any antibody so we labeled them as seronegative. Seronegative MG carried a vivid relation with earlier onset (p=0.07) as 94.4% of seronegative and 62% of seropositive patients developed MG before 35 years of age. In gender distribution 71.7% males and 89% females were seropositive and interestingly MuSK-Ab were seen only in females (Table 1).
| Age of onset (Years) |
Acetylcholine Receptor Ab |
MuSK Ab |
Sero-negative |
Total |
| M |
F |
M |
F |
M |
F |
| ≥20 y |
1 |
21 |
-- |
1 |
1 |
5 |
29 |
| 21-35 y |
8 |
16 |
-- |
4 |
9 |
3 |
40 |
| 35-50 y |
9 |
10 |
-- |
2 |
-- |
-- |
21 |
| >50 y |
10 |
1 |
-- |
-- |
1 |
-- |
12 |
| Total |
28 |
49 |
-- |
7 |
11 |
7 |
102 |
Table 1: Showing demographic details of the MG patients sorted according to various auto- antibodies.
In general, seropositive patients represented relatively severer form of MG on MGFA grading (p=0.02) independent of any confounding factors, especially MuSK-Ab MG had comparatively stronger manifestations () than AchR-Ab positive MG. Seropositivity was noticeably observed more in general form of MG (p=0.001).
Myasthenia crisis (MC) is well known entity, which represents acute severe worsening of clinical features, which might need rescue immunomodulation and hospitalization. From our patients 60 (58.8%) experienced crisis and out of these 17 (28.3%) had frequent and severe crisis (>3 times) in 5 years follow up. As a result, we encountered 141 crisis accumulatively. In terms of Immunology 67% of AchR-Ab positive patients, all of MuSK-Ab positive and only 33% of seronegative patients underwent crisis, hence crisis occurred predominantly in seropositive myasthenia (p<0.05). A mention-worthy observation indicated that severer the MG at start (Osserman’s class>3) frequent are the crisis events (p=0.046).
We entertained the frequency and severity of crisis on demographic parameters too and did not find any genderbased trend, however ethnicity wise Arab population had more frequent crisis (p=0.04). But it might not be a true predictor as majority of expats discontinued follow up while repatriated, owing to which we might have missed some data. Age of MG onset or current age of patients did not establish any particular association to frequency or gravity of crisis, however it was noticed that patients with seronegative MG usually develop crisis at relatively late age (Figure 1). Average time from diagnosis to first crisis was 3.56 years, it was earlier in AchR-Ab MG in comparison to Anti-MuSK+ MG. Duration after the diagnosis did not impact enormously the degree of MG crisis or its frequency.
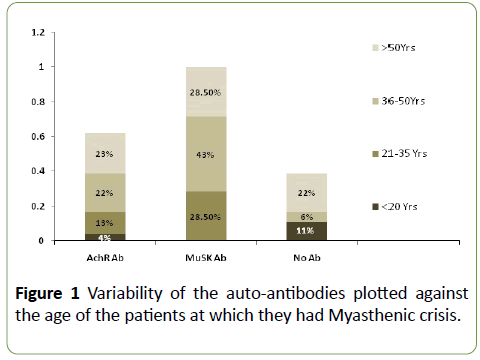
Figure 1: Variability of the auto-antibodies plotted against the age of the patients at which they had Myasthenic crisis.
Severity of each crisis was calibrated in terms of Myasthenia gravis foundation of America (MGFA) grading and it was noticed that 31.6% crisis were of milder degree and 68% were of moderate to severe nature. An interesting point appeared here that frequency of MG crisis in various patients showed a direct proportion to crisis intensity on MGFA scale i.e., the patients who had more frequent crisis they experienced relatively severer crisis (p=0.007). Seropositive MG patients had about 30% more tendency to develop crisis (p=0.019), especially patients with MuSK-Ab MG had more frequent crisis, however there was no statistically significant relationship between severity of crisis with any particular antibodies. Respiratory instability is one of the most concerned elements during a crisis which might lead to intensive care admission and mechanical ventilation and it was observed predominantly in seropositive MG patients (p=0.03).
Though there was no statistically mention-worthy relationship of crisis intensity to any particular antibodies, however anti-MUSK-Ab MG showed relatively severer crisis as depicted in Table 2.
| Variables |
I |
II a |
II b |
III a |
III b |
IV a |
IV b |
V |
Total |
| Acetylcholine Receptor Ab |
2 |
18 |
18 |
18 |
12 |
5 |
12 |
13 |
98 |
| MuSK Ab |
-- |
-- |
9 |
3 |
6 |
3 |
3 |
3 |
28 |
| Sero-negative |
-- |
3 |
6 |
4 |
1 |
1 |
-- |
-- |
15 |
| Total |
2 |
21 |
33 |
25 |
19 |
9 |
16 |
16 |
141 |
Table 2: Severity of Myasthenia crisis on Myasthenia Gravis Foundation of America (MGFA) scale sorted in relation to MG auto-antibodies.
All the patients were necessarily on baseline optimal therapy for MG. None of them was without treatment during the period of data collection, however poor compliance was a noticeable trigger factor in few of the patients. Rest of the predisposing factors included infection, surgery and obstetric causes especially postpartum periods. On average AchR-Ab patients experienced 1.2 crisis per patient, Anti-MuSK-Ab had 4 crises per patient and seronegative patients showed 0.82 crisis per patient. Average duration of hospital stay during crisis was relatively longer for MG with AchR-Ab patients (Table 3) but antibody type seems unlikely to predict the hospital course independently, as many contributors including infection, DVT, co-morbidities and trigger factor of crisis might have played their role in prolonged hospital stay.
| Clinical Parameters |
Acetylcholine Recep Ab MG |
muscle-specific Tyrosine kinase, (MuSK) MG |
Seronegative MG |
| Gender |
Male |
28 (36%) |
0 |
11 (61%) |
| Female |
49 (64%) |
7 (100%) |
7 (39%) |
| Current Age |
≤ 35 years |
25 (32.5%) |
2 (28.5%) |
8 (44.4%) |
| >35 years |
52 (67.5%) |
5 (72.5%) |
10 (55.6%) |
| Age (At onset) |
= 35 years |
47 (61%) |
5 (72.5%) |
17 (94.4%) |
| >35 years |
30 (39%) |
2 (28.5%) |
1 (5.6%) |
| Ethnicity |
Arabs |
58 (75%) |
7 (100%) |
17 (94.4%) |
| Non-Arabs |
19 (24.6%) |
0 |
1 (5.6%) |
| Types MG |
Ocular |
7 (9%) |
0 |
10 (55.6%) |
| General |
70 (91%) |
7 (100%) |
8 (44.4%) |
| Osserman’s class |
1-3 |
72 (93.5%) |
6 (85.7%) |
18 (100%) |
| 4-5 |
5 (6.5%) |
1 (14%) |
0 |
| Clinical Distribution |
Ocular Symptoms |
60 (77.9%) |
6 (85.7%) |
18 (100) |
| Bulbar symptoms |
45 (58.4%) |
7 (100%) |
4 (22.2%) |
| Motor symptoms |
61 (79%) |
5 (71.4%) |
11 (61%) |
| Respiratory symp |
13 (17%) |
2 (28.5%) |
0 |
| ICU care/MV |
21 (27%) |
3 (42.8%) |
1 (5.5%) |
| MG crisis |
47 (61%) |
7 (100%) |
6 (33%) |
| MGFA grade of worst crisis |
I/IIa/IIb |
14 (29.7%) |
2 (28.7%) |
3 (50%) |
| IIIa/IIIb/IVa/IVb/V |
33 (70.3%) |
5 (71.3%) |
3 (50%) |
| Crisis/pt (Average) |
1.2 |
4.0 |
0.82 |
| Hospital stay (Average in days) |
11.7 |
10.2 |
7.4 |
| Thymic Pathology |
31 (56%) |
0 |
5 (31%) |
| Autoimmune disease |
10 (12.9%) |
0 |
2 (11%) |
| Neoplasm |
4 (5%) |
2 (28.5%) |
0 |
| Thyroid disease |
11 (14.2%) |
0 |
3 (16.6%) |
Table 3: Description of various demographic and clinical parameters of MG patients, according to auto-antibody distribution.
One of our patients expired during crisis, so our mortality rate is 1.6%. He had AchR-Ab positive MG and presented with crisis of MGFA class V. Extended hospital stay and Poor outcome were related to respiratory failure, pneumonia, other infections, elderly age and higher MGFA/Osserman’s grading of MG at onset.
As origin of myasthenia is pathologically relates to thymus, we entertained thymus pathology as well indicating presence of thymoma in 17.6% cases, thymic hyperplasia in 17.6%, whereas 38.2% patients were normal and in 26.5% no investigation for thymus was carried out. Surprisingly, presence of auto-antibodies failed to establish a statistically significant relation with thymic pathology, however thymectomy showed a meticulous response and that was also classified in terms of autoantibodies. A total of 65% patients underwent thymectomy and average duration between diagnosis and thymectomy was 2.5 years. The data highlighted a significant benefit in form of decline in frequency and severity of crisis and rest of parameter, following thymectomy. Utmost benefit was seen in patients with AchR Ab MG whereas patients with Anti-MuSK+ MG did not experience any benefit post thymectomy (p=0.01). Earlier thymectomy after diagnosis indicated a better prognosis but statistically strong association between duration and outcome was lacking (p=1.3).
As MG is an autoimmune disorder and its coexistence with other autoimmune disorders is not a wonder but presence of neoplasm and certain other disorders might play a role of foundation stone for further researches. We noticed presence of neoplasm in 6 (4.2%), autoimmune disorders other than myasthenia in 12 (8.5%) and thyroid disorder in 14 (10%) of our patients (Table 3).
Neoplasm including Leiomyosarcoma, Retroperitoneal sarcoma, Malignant thymoma, uterine fibroids and Thyroid carcinoma were found and patients with all neoplasms encountered severe degree of MG except those with Leimyosarcoma and Fibroids, who improved after the resection of tumors. Immune disorders included Hashimoto's thyroiditis, SLE, Mixed connective tissue disorder and Bachet's disease. MGFA grading/crisis in such patients graded at an average degree.
No mentionable relation of coexistent neoplasm, other autoimmune disorders, thyroid disease and thymic pathology was ascertained to autoantibody type and MG crisis (Table 3).
Discussion
Like all other autoimmune diseases, myasthenia gravis affects Caucasians more than other ethnicities [6]. In a regional Saudi cohort patient of Myasthenia were relatively young, onset peak was in second and third decades for females, whereas it was in third and fourth decade for males [9]. This is in accordance to our findings.
Approximately 15-20% of patients with MG experience crisis in their lifetime, typically within the early years of the diagnosis [10]. Thomas et al. has found a median time of 8 to 12 months of first myasthenia crisis from onset of symptoms [11]. Myasthenic crisis may be the initial presentation of MG in 20% of patients [12].
Overall, women are twice as likely as men to be affected by MC and similar ratio is endorsed by our data [13]. A bimodal distribution of MC is witnessed, an early peak predominant in females and a later peak after age 55 affects women and men equally [13]. Panda et al. highlighted that the subset of Indian patients with MG who are at risk to develop MC belong to the 3rd and 4th decade of their life, such patients predominantly have bulbar symptoms at presentation and are likely associated with thymoma [14-24].
In 80-85% cases of MG, antibodies against the skeletal muscle nicotinic acetylcholine receptors are responsible for clinical implication and in 10-15% of the rest Anti-MuSK-Ab has been identified [1]. Though serum assays do not reflect the actual quantity of antibodies attached to motor endplate, yet it is believed that higher antibody titers indicate disease of severe degree [14]. There are several observations indicating that the AChR-Ab is directly responsible for the clinical symptoms such as a decrease in the number of active acetylcholine receptors has been demonstrated to occur as a consequence of AChR antibody binding [14]. This may be due to autoantibody binding alone early in the disorder, but destruction of receptors eventually occurs via a complementmediated process [15].
However, the linkage between AChR-Ab titer and MG is not absolute. The plasma concentration of AChR-Ab does not correlate well with the severity of the disease. A low-titer or even antibody-negative patient may have much more severe clinical disease than a patient with high titers. However, still few researchers believe that titers tend to fall with successful immunotherapy, and they go parallel with clinical improvement [15].
In 2012 in a Japanese cohort of MG auto-antibodies against the main immunogenic region (MIR) located at the extracellular end of the nicotinic acetylcholine receptor α- subunit were checked and their clinical relevance in MG was studies. This indicated positive association of antibody titer with disease severity, bulbar symptoms and thymoma, and negative association with ocular MG [16]. We did not study the antibody titer impact on clinical parameters of disease; however, we studied various antibodies and their effect on disease course, and these seem to play pretty important role in disease pattern.
Anti-MuSK antibodies have been reported in 65-71% of generalized MG patients who were negative for AchR Ab [17]. Such patients clinically exhibited severe oculobulbar weakness and responded poorly to treatment as per Scuderi et al. [18]. MuSK-Ab positive myasthenia may have a different cause and pathologic mechanism than AChR-antibody positive disease [19]. In contrast to AChR-Ab-positive MG, in which complement fixing IgG1 and IgG3 subclasses predominate [20], in MuSK-Ab IgG4 are mainly potent [21]. However, there is evidence that IgG1 subclass antibodies are also present at a low concentration and are capable of activating complement when bound to MuSK receptor [22]. This may account for some potential mechanism involved in MG pathology rendering clinical variance.
Sanders et al. categorized Anti MuSK MG into three distinct clinical presentations: one that is indistinguishable from anti- AChR Ab MG; second with severe oculobulbar weakness; and third with predominantly neck, shoulder, and respiratory muscle weakness, with little or delayed ocular muscle weakness, whereas response to treatment was reportedly variable [17]. Guptill et al. reviewed 110 MuSK-Ab MG patients and concluded that 85% were MGFA class III or above and crisis occurred in 28% [23]. Similarly, our data indicated relatively severer form of MG (Osserman’s class ≥ 3) with Anti MuSK-Ab and these patients faced relatively frequent and severer crisis.
One third of the patients who survive the first crisis experience a second crisis. Myasthenic crisis is more common in patients of myasthenia gravis associated with thymoma [24]. Similarly, our data indicated presence of crisis in 83.5% of patients with thymoma, 55% patients with thymus hyperplasia and 46% of those with normal thymus.
For MG mortality rate in hospitals in the US is 2.2%, and it is increased to 4.4% in MG with crisis. [6] Whereas our mortality rate during crisis is 1.6%. The survival rates of MG after diagnosis at year 3 were 85%, at year 5 were 81%, at year 10 were 69%, and at year 20 were 63% in Denmark [25].
A detailed analysis of correlation between antibodies and disease severity and crisis is probably not studied before, so we studied a comparison of various antibodies in terms of their impact crisis. Currently available information depicts seropositive MG bears a worse disease pattern and amongst seropositive Anti-MuSK MG carries severer clinical parameters with frequent crisis’ and relatively poor prognosis. Above described debate enlightens that titer of antibodies might not represent the severity of MG course with certainty, however presence or absence of various antibodies might show prognostic trends. Further work in this context may disclose more facts in the future.
Conclusion
MG is autoimmune post-synaptic neuromuscular disorder majorly afflicting females with M:F ratio of 1:1.6. Age of onset is relatively younger for females. A high MGFA grade at presentation predicts higher probability of crisis.
Seropositive MG is usually severe form of Myasthenia and further in this group MuSK-Ab MG showed higher MGFA grading at presentation and more frequent MG crisis than AchR-Ab MG. Patients who suffered more frequent crisis, their crisis were qualitatively stronger on MGFA scale, as well. Average hospital stay was relatively longer for AchR-Ab positive patients.
Bulbar weakness was seen more frequent in seropositive patients and mechanical ventilation was required more in this group as well. Seronegative MG has usually earlier onset, milder disease and better prognosis.
Conflict of Interest
None of the authors has any disclosure or conflict of interest.
Acknowledgment
Dr Maria Khan Junaidi, FCPS Neurology and Biostatistics expert. She gave us idea about analysis of our data.
Funding
This study was not funded by any authority.
24128
References
- Meriggioli MN (2009) Myasthenia Gravis: Immunopathogenesis, diagnosis and management. Continuum 15: 35-62.
- Conti-Fine BM, Milani M, Kaminski HJ (2006) Myasthenia gravis: Past, present, and future. J Clin Invest 116: 2843-2854.
- Juel VC, Massey JM (2007) Myasthenia gravis. Orphanet J Rare Dis 2: 44.
- Illa I, CortÃÂÃÂÃÂâÂÂÃÂâ â≢ÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂés-Vicente E, MartÃÂÃÂÃÂâÂÂÃÂâ â≢ÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂnez MÃÂÃÂÃÂâÂÂÃÂâ â≢ÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂÃÂ, Gallardo E (2018) Diagnostic utility of cortactin antibodies in myasthenia gravis. Ann NY Acad Sci 1412: 90-94.
- Sieb JP (2014) Myasthenia gravis: An update for the clinician. Clin Exp Immunol 175: 408-418.
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ (2009) Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 72: 1548-1554.
- Aydin Y, Ulas AB, Mutlu V, Colak A, Eroglu A (2017) Thymectomy in Myasthenia Gravis. Eurasian J Med 49: 48ÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâââÂÂìÃÂ
âÂÂ52.
- Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, et al. (2016) International consensus guidance for management of myasthenia gravis. Neurology 87: 419-425.
- Al-Moallem MA, Alkali NH, Hakami MA, Zaidan RM (2008) Myasthenia gravis: Presentation and outcome in 104 patients managed in a single institution. Ann Saudi Med 28: 341-345.
- Bedlack RS, Sanders DB (2002) On the concept of myasthenic crisis. J ClinNeuromuscul Dis 4: 40-42.
- Wendelland LC, Levine JM (2011) Myasthenic crisis. Neurohospitalist 1: 16-22.
- Rabinstein AA, Mueller-Kronast N (2005) Risk of extubation failure in patients with myasthenic crisis. Neurocrit Care 3: 213-215.
- Thomas CE, Mayer SA, Gungor Y, Swarup R, Webster EA, et al. (1997) Myasthenic crisis: Clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology 48: 1253-1260.
- Drachman DB, Adams RN, Josifek LF, Self SG (1982) Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 307: 769-775.
- Conti-Fine BM, Milani M, Kaminski HJ (2006) Myasthenia gravis: Past, present, and future. J Clin Invest 116: 2843-2854.
- Masuda T, Motomura M, Utsugisawa K, Nagane Y, Nakata R, et al. (2012) Antibodies against the main immunogenic region of the acetylcholine receptor correlate with disease severity in myasthenia gravis. J Neurol Neurosurg Psychiatry 83: 935-940.
- Sanders DB, El-Salem K, Massey JM, McConville J, Vincent A (2003) Clinical aspects of MuSK antibody positive seronegative MG. Neurology 60: 1978-1980.
- Scuderi F, Marino M, Colonna L, Mannella F, Evoli A, et al. (2002) Anti-P110 autoantibodies identify a subtype of ÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâ¦ââ¬Ã
ÂseronegativeÃÂÃÂÃÂâÂÂÃÂâÂÂÃÂâÃÂÃÂââ¬Ã
¡ÃÂâââ¬Ã
¡ÃÂìÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂàmyasthenia gravis with prominent oculobulbar involvement. Lab Invest 82: 1139-1146.
- McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, et al. (2004) Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 55: 580-584.
- RÃÂÃÂÃÂâÂÂÃÂâ â≢ÃÂÃÂââ¬Ã
¡ÃÂâÂÂÃÂødgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S (1987) Acetylcholine receptor antibody in myasthenia gravis: Predominance of IgG subclasses 1 and 3. Clin Exp Immunol 67: 82-88.
- McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, et al. (2004) Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 55: 580-584.
- Leite MI, Jacob S, Viegas S, Cossins J, Clover L, et al. (2008) IgG1 antibodies to acetylcholine receptors in 'seronegative' myasthenia gravis. Brain 131: 1940-1952.
- Guptill JT, Sanders DB, Evoli A (2011) Anti-MuSK antibody myasthenia gravis: Clinical findings and response to treatment in two large cohorts. Muscle Nerve 44: 36-40.
- Panda S, Vinay G, Behari M, Singh S, Srivastava T (2004) Myasthenic crisis: A retrospective study. Neurology India 52: 453-456.
- Carr AS, Cardwell CR, McCarron PO, McConville J (2010) A systematic review of population-based epidemiological studies in myasthenia gravis. BMC Neurol 10: 46.

