Keywords
Epilepsy; Neurological disorder; Pathophysiology
Introduction
Epilepsy affects approximately 42 million people worldwide and is the most common heterogeneous neurological disorder with distinct symptoms, etiology, prognosis and treatments [1]. Cases of epilepsy may be organized into distinct epilepsy syndromes by the specific electro clinical features that are present. JME is a common form of idiopathic generalized epilepsy which represents 5-10% of all epilepsy cases [2]. Genetic studies have demonstrated at least 6 loci for JME and most of which are ion channels genes [3]. Recent progress in genome and exome sequencing has revealed that some individuals diagnosed with LGS have de novo mutations in a variety of genes [4]. Nearly 70% of patients with epilepsy lack an obvious pathogenetic cause and genetics is believed to play an important role in its causation. Mutations/polymorphisms in nicotinic acetylcholine receptor genes, GABA receptor gene, voltage-gated sodium channel genes etc. may contribute to pathogenesis of epilepsy [5-7]. Juvenile Myoclonic Epilepsy (JME) also known as Janz syndrome is a condition characterized by recurrent seizures [8]. The onset symptoms usually occur between 12 and 18 years of age and it lasts into adulthood. The most common type of seizures in JME is myoclonic seizures and these patients may also have generalized tonic-clonic seizures (GTCS) [9]. Generally, patients with JME have the characteristic myoclonic seizures in adolescence, which after few years develop into GTCS. The genetics of JME is complex and has not been understood completely [3]. Mutations in genes related to neurotransmission are believed to increase the susceptibility to this condition. GABRA1 and EFHC1 genes which encode GABA receptor α1 subunit and EF hand containing protein 1 respectively are the most studied in JME but many JME patients do not have mutations in any of these genes [10]. A number of still unidentified genes are likely to be involved in pathogenesis of JME [11]. Lennox-Gastaut syndrome (LGS) is a form of severe epilepsy that begins in childhood [12]. It may occur as a result of an insult to brain during foetal life or soon after birth, or it may manifest in a previously normal child. It is characterized by multiple types of seizures including tonic, atonic and atypical absences [13].
The onset of seizure in LGS is usually in early childhood, between 3 and 5 years of age. In most cases, LGS patients usually suffer from intellectual disability as well. More than three-quarters of affected individuals with LGS have tonic seizures while atypical seizures are also common. No genes specific to LGS have been identified, although clinical observations suggest that the disorder is likely to have a genetic component [14].Voltage-gated sodium channels play an important role in action potential generation and membrane excitability [15]. Genes coding for sodium channel components are considered to be one of the most important and major class of genes associated with various epilepsy phenotypes [16]. The voltage-gated sodium ion channels consist of large α subunits that associate with other polypeptides, such as β subunits to form functional voltagegated ion channels [17]. The alpha subunit of sodium channel is a highly processed approximately 260-kDa polypeptide, that is comprised of four homologous domains termed I–IV [18]. Within each domain, there are six transmembrane segments called S1-S6 [19]. A hairpin-like P-loop between S5 and S6 forms part of the channel pore, and the intracellular loop that connects domains III and IV forms the inactivation gate [20]. Initiation and propagation of seizures is due to misfiring of neurons in the brain, and >300 mutations in SCN1A gene have been identified in epilepsy and other neurological disorders so far [21]. There are multiple alleles of the alpha subunit gene referred to as SCN1A through SCN11A [22]. The individual sodium channels are distinguished not only by differences in their sequence but also by their expression profiles and kinetics. Alterations in SCN1A gene are one of the major causes of Dravet syndrome [16]. SCN1A variants can result in a wide spectrum of epilepsy phenotypes, ranging from severe epilepsies, such as intractable childhood epilepsy with generalized tonic-clonic seizures (ICE-GTC), to milder epilepsy disorders, including simple febrile seizures or familial feverrelated epilepsies referred to as generalized/genetic epilepsies with febrile seizures plus [23]. More recent studies have indicated that common variants in the SCN1A gene may be risk factors for common epilepsies like temporal lobe epilepsy and idiopathic/genetic generalized epilepsy (GGE/IGE) [24]. Alterations in SCN2A are one of the most common causes of neurodevelopmental disease and phenotypes include autism/ intellectual disability/schizophrenia, infantile spasms progressing to epileptic encephalopathy and severe earlyonset epileptic encephalopathy [25-27].
Defects in subunits of sodium channels render them susceptible to slow inactivation, i.e., the membrane remains depolarized for a longer time, which can result in epileptogenesis and spread of seizures [28,29]. Altered sodium channel transcript levels had been found in brain tissues of epilepsy patients, suggesting a potential role for sodium channels in the pathophysiology of epilepsy [30]. In the last few decades, various coding and non-coding sequence variations in genes of voltage-gated sodium channels i.e., SCN1A, SCN2A, SCN8A and SCN9A have been identified in patients with seizures, ataxia, and sensitivity to pain [31]. Though various single nucleotide polymorphisms (SNPs) in sodium channel genes have been described so far, only a few including SCN1A Thr1067Ala i.e., 3184 A>G (rs2298771) and SCN2A Arg19Lys i.e., 56 G>A (rs17183814), were found to have functional significance in different neurological disorders [32]. The Arg19Lys polymorphism and other missense mutations in SCN2A gene were found in a Japanese family that had febrile seizures (FS) [33]. The SCN1A Thr1067Ala polymorphism results in change of amino acid threonine to alanine at a highly consensus conserved site in the coding region of the SCN1A, which might possibly affect the functioning of inactivation gate of sodium channel in the cytosol that regulates efflux and influx of sodium ions [21]. Another possibility is that this polymorphism is in linkage disequilibrium with some other genetic variants of the same gene that impart increased risk for developing epilepsy. Defects in SCN1A gene might lead to myoclonic epilepsy in infancy (SMEI), generalized epilepsy with febrile seizures (GEFS) plus type 2, and intractable childhood epilepsy with generalized tonic-clonic seizures [34,35]. The Arg19Lys polymorphism causes amino acid substitution in cytoplasmic portion of the sodium channel and is a moderately conserved residue. The SCN2A Arg19Lys variant that codes for lysine was found to be significantly more frequent in patients with Febrile Seizures (FS) including GEFS than in controls [36]. Along with other genetic variants, SCN2A Arg19Lys polymorphism Lys allele may play an important role in epilepsy susceptibility. The causal relationship between SCN1A Thr1067Lys and SCN2A Arg19Lys polymorphisms with epilepsy syndromes (like JME and LGS) has not been proven conclusively. Analysis of association of these polymorphisms with epilepsy syndromes in different populations would be important in establishing a role for SCN1A and SCN2A genes in the development of seizures. The main aim of this study was to evaluate the role of SCN1A Thr1067Ala and SCN2A Arg19Lys gene polymorphisms in the pathophysiology of JME and LGS in North Indian population.
Methodology
Study population
A total of 50 JME patients, 50 LGS Patients and 100 age and sex matched healthy volunteers were recruited in this study. Clinical data from each patient with reference to frequency and duration of seizures, postictal period, drug responsiveness, age of onset of seizure etc. was collected and analysed. JME patients were diagnosed by a combination of clinical and EEG criteria. The clinical criteria for JME included presence of generalized tonic clonic seizures (GTCS) and normal cognition. The EEG criteria for JME included generalized spike wave discharge (4-6 Hz), polyspike wave discharge and photo paroxysmal response. JME patients with seizure type as only myoclonic Jerks and GTCS were recruited in order to make the study homogenous
LGS patients were also diagnosed by a combination of clinical and EEG criteria. The clinical criteria for LGS included presence of multiple seizure types and presence of cognitive impairment. The EEG criteria for LGS included multifocal spike wave discharge (<2.5 Hz), slow spike wave discharge and generalized paroxysmal fast activity (GPFA). LGS patients having tonic seizures as the main seizure type were recruited in order to make the study homogenous. Patients with epilepsy syndromes other than JME and LGS were excluded from the study. The study was approved by institutional ethics committee, Maulana Azad Medical College (IEC-MAMC), New Delhi, India.
Peripheral blood sample collection.
Peripheral blood samples (3 ml) were collected in EDTA vials from patients and healthy control subjects. Genomic DNA was isolated from peripheral blood leukocytes using standard phenol chloroform DNA extraction method.
Genotyping of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphism
All samples were genotyped for SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms using polymerase chain reaction-restriction fragment length polymorphism- (PCRRFLP) method. DNA fragments containing these polymorphic sites were amplified by PCR with specific forward and reverse primers in a final volume of 25 μl containing 50-100 ng genomic DNA as shown in Table 1.
| SNP |
Sequence |
Primer Sequence |
Restriction Enzyme |
| SCN1A Thr1067Ala |
Forward |
5 -TGCACAAAGGAGTAGCTTATG-3 |
PvuII |
| Reverse |
5 -AGTCAAGATCTTTCCCAATTTCAG-3 |
| SCN2A Arg19Lys |
Forward |
5 -AATCACCTTTTATTCTAATGGTC-3 |
Bme1390I |
| Reverse |
5 -CAGTGAAGGCAACTTACTAAGA-3 |
Table 1: Primers for SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms.
PCR conditions included: a denaturing step at 95°C for 5 min, then 30 cycles of 94°C for 30 s, 60°C (SCN2A Arg19Lys)/ 57°C (SCN1A Thr1067Ala) for 30 s, 72°C for 30 s, and a final incubation at 72°C for 10 min. After amplification, PCR products were digested using specific restriction endonucleases. The SCN1A Thr1067Ala polymorphism genotypes were identified by presence/loss of PvuII restriction site (A allele, 168 bp; G allele, 145 and 23 bp) as shown in Figure 1. The SCN2A Arg19Lys polymorphism genotypes were identified by presence/loss of the Bme1390I restriction site (G allele 178, 130, 64 and 28 bp; and an allele 206,130 and 64 bp) as shown in Figure 2.
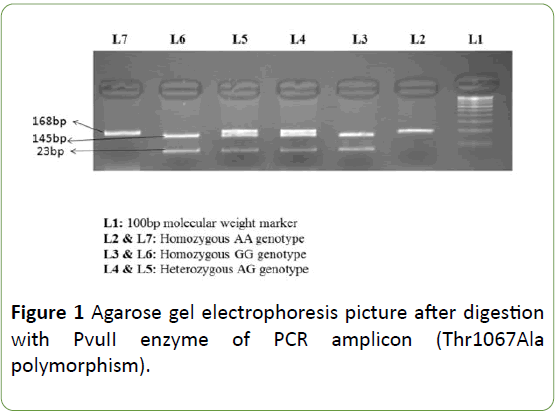
Figure 1: Agarose gel electrophoresis picture after digestion with PvuII enzyme of PCR amplicon (Thr1067Ala polymorphism).

Figure 2: Agarose gel electrophoresis picture after digestion with Bme1390I enzyme of PCR amplicon (Arg19Lys polymorphism).
Statistical analysis
All statistical analyses were performed using Graph Pad prism version 7.03. Chi square and fisher exact test were used to estimate the statistical significance of differences observed between various groups. Risk of epilepsy syndromes associated with inheritance of different genotypes was calculated by odds ratio. P-value of <0.05 was considered significant.
Results
50 JME, 50 LGS patients and 100 age and sex matched healthy control subjects were included in the study.
Demographic features of the patients including age, gender and family history of epilepsy are shown in Tables 2 and 3. In JME group, there were 20 male and 30 female subjects while in LGS group there were 42 male and 8 female subjects. Based on the median age, JME patients were divided in two age groups: patients with age ≤ 20 years included 29 cases and >20 years included 21 cases. Similarly, LGS patients were divided in two age groups: patients with age ≤ 7 years included 24 cases and those >7 years included 26 cases.
| Variables |
JME patients |
| Number of Patients |
50 |
| Gender |
| Male |
20 |
| Female |
30 |
| Age |
| ≤ 20 years |
29 |
| >20 years |
21 |
| Family history of epilepsy |
| Yes |
10 |
| No |
40 |
Table 2: Demographic characteristics of Juvenile myoclonic epilepsy (JME) patients.
| Variables |
LGS patients |
| Number of Patients |
50 |
| Gender |
| Male |
42 |
| Female |
8 |
| Age |
| ≤ 7 years |
24 |
| >7 years |
26 |
| Family history of epilepsy |
| Yes |
10 |
| No |
40 |
Table 3: Demographic characteristics of Lennox-Gastaut syndrome (LGS) patients.
Comparative analysis of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphism
The comparative analysis of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphism genotypic and mutant/wild allelic distribution is shown in Tables 4 and 5.
| SNPs |
JME N=50 (%) |
LGS N=50 (%) |
Controls N=100 (%) |
p value |
| cases vs. controls |
JME vs. LGS |
| JME |
LGS |
| SCN1A Thr1067Ala |
| AA |
34 (68) |
42 (84) |
76 (76) |
0.4 |
0.008 |
0.0008 |
| AG |
5 (10) |
6 (12) |
7 (7) |
| GG |
11 (22) |
2 (4) |
17 (17) |
| SCN2A Arg19Lys |
| GG |
43 (86) |
42 (84) |
89 (89) |
0.27 |
0.03 |
0.54 |
| GA |
4 (8) |
6 (12) |
3 (3) |
| AA |
3 (6) |
2 (4) |
8 (8) |
Table 4: Comparative analysis of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphism genotypic distribution in JME and LGS cases and healthy control subjects.
| SNPs |
JME (N=50) |
LGS (N=50) |
Controls (N=100) |
p value |
| cases vs. controls |
JME vs. LGS |
| JME |
LGS |
| SCN1A Thr1067Ala |
| A |
0.73 |
0.9 |
0.8 |
0.32 |
0.04 |
0.002 |
| G |
0.27 |
0.1 |
0.2 |
| SCN2A Arg19Lys |
| G |
0.9 |
0.9 |
0.9 |
1 |
1 |
1 |
| A |
0.1 |
0.1 |
0.1 |
Table 5: Comparative analysis of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphism allelic distribution in JME and LGS cases and healthy control subjects.
Compliance with Hardy-Weinberg equilibrium was ascertained amongst the control subjects.
SCN1A Thr1067Ala polymorphism genotypic and allelic distribution in JME & LGS patients and control subjects
JME vs. healthy control subjects: Though the percentage of mutant heterozygous (AG) and mutant homozygous (GG) genotypes was higher in JME cases than in controls, there was no significant difference in the genotypic and mutant/wild allelic distribution (p=0.4 and 0.32 respectively) of SCN1A Thr1067Ala polymorphism amongst JME cases versus controls.
LGS vs. healthy control subjects: There was significant difference in the genotypic and mutant/wild allelic distribution (p=0.008 and 0.04 respectively) of SCN1A Thr1067Ala polymorphism amongst LGS cases versus controls. The percentage of mutant heterozygous (AG) plus mutant homozygous (GG) genotypes (16%) was found to be lower in LGS cases than in controls (24%) as shown in Table 4. The mutant G allele frequency in LGS patients (0.1) was half of that in the control subjects (0.2) as shown in Table 5.
JME vs. LGS: There was a significant difference in the genotypic and mutant/wild allelic distribution (p=0.0008 and 0.002 respectively) of SCN1A Thr1067Ala polymorphism amongst JME cases versus LGS cases. The percentage of mutant heterozygous (AG) plus mutant homozygous (GG) genotypes was much lower in LGS cases (16%) than in JME cases (32%) as shown in Table 4. The mutant G allele frequency in LGS patients (0.1) was nearly 1/3rd of the same in JME patients (0.27) as shown in Table 5.
SCN2A Arg19Lys polymorphism genotypic and allelic distribution in JME & LGS patients and control subjects
JME vs. healthy control subjects: There was no significant difference in genotypic and mutant/wild allelic distribution (p=0.27 and 1 respectively) of SCN2A Arg19Lys polymorphism amongst JME cases versus controls. The percentage of mutant heterozygous (GA) genotype was found to be higher in JME cases than in controls, while the percentage of mutant homozygous (AA) genotype was lower in JME cases than in controls as shown in Table 4.
LGS vs. healthy control subjects: There was significant difference in genotypic distribution (p=0.03) but no significant difference in mutant/wild allelic distribution (p=1) of SCN2A Arg19Lys polymorphism amongst LGS cases versus controls. In LGS cases, the percentage of mutant heterozygous (GA) genotype was higher while the percentage of mutant homozygous (AA) genotype was lower than in controls as shown in Table 4.
JME vs. LGS: There was no significant difference in genotypic and mutant/wild allelic distribution (p=0.54 and 1 respectively) of SCN2A Arg19Lys polymorphism amongst JME cases as compared to that in LGS cases.
SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms and risk of epilepsy syndromes
Evaluation of susceptibility to JME and LGS associated with inheritance of mutant genotypes of SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms was done by computation of odds ratio (Tables 6 and 7).
| Genotypes |
Cases n=50 (%) |
Controls n=100 (%) |
OR (95% CI) |
p value |
| JME |
AA |
34 (68) |
76 (76) |
Ref |
- |
| AG |
5 (10) |
7 (7) |
1.59 (0.58-4.43) |
0.45 |
| GG |
11 (22) |
17 (17) |
1.45 (0.71-2.95) |
0.37 |
| LGS |
AA |
42 (84) |
76 (76) |
Ref |
- |
| AG |
6 (12) |
7 (7) |
1.55 (0.58-4.14) |
0.47 |
| GG |
2 (4) |
17 (17) |
0.21(0.07-0.66) |
0.005 |
Table 6: SCN1A Thr1067Ala polymorphism and associated risk of epilepsy syndromes.
| Genotypes |
Cases n=50 (%) |
Controls n=100 (%) |
OR (95% CI) |
p value |
| JME |
GG |
43 (86) |
89 (89) |
Ref |
- |
| GA |
4 (8) |
3 (3) |
2.76 (0.71-10.75) |
0.21 |
| AA |
3 (6) |
8 (8) |
0.78 (0.26-2.3) |
0.79 |
| LGS |
GG |
42 (84) |
89 (89) |
Ref |
- |
| GA |
6 (12) |
3 (3) |
4.24 (1.15-15.55) |
0.029 |
| AA |
2 (4) |
8 (8) |
0.53 (0.15-1.83) |
0.38 |
Table 7: SCN2A Arg19Lys polymorphism and associated risk of epilepsy syndromes.
SCN1A Thr1067Ala polymorphism mutant homozygous GG genotype was associated with a significantly lower risk of LGS as compared to the reference wild type AA genotype with an odds ratio of 0.21 (95% CI 0.07 to 0.66, p=0.005).
An increased risk of LGS with odds ratio of 4.24 (95% CI 1.15 to 15.55, p=0.029) was observed in association with mutant heterozygous GA genotype of SCN2A Arg19Lys polymorphism; however, the same was not observed with mutant homozygous genotype AA genotype. No alteration in susceptibility to JME was noted with either SCN1A Thr1067Ala or with SCN2A Arg19Lys polymorphism.
Clinical features of JME and LGS patients and SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms
Differential analysis of clinical details of patients such as age at onset of seizures, frequency and duration of seizures, drug responsiveness etc. with respect to SCN1A Thr1067Ala and SCN2A Arg19Lys are shown in (Tables 8-11). Patients who showed a decrease in the frequency/severity of seizures after drug treatment were classified as drug responsive and those who did not as non-drug responsive. Mean age of onset of seizures in JME patients was 13.31 ± 4.54 years (Range: 2.5 -22 years), while the mean age of onset of seizures in LGS patients was 2.06 ± 2.818 years (Range: 1-13 years).
| Variables |
JME patients (%) |
AA |
AG |
GG |
p value |
A allele frequency |
G allele frequency |
| Onset of seizures |
| ≤ 15 years of Age |
34 (68) |
26 (76.5) |
2 (6) |
6 (17.5) |
0.15 |
0.79 |
0.21 |
| >15 years of Age |
16 (32) |
8 (50) |
3 (19) |
5 (31) |
0.59 |
0.41 |
| Frequency of seizures |
| ≤ 1 per month |
21 (42) |
12 (57) |
3 (14.5) |
6 (28.5) |
0.35 |
0.64 |
0.36 |
| >1 per month |
29 (58) |
22 (76) |
2 (7) |
5 (17) |
0.79 |
0.21 |
| Duration of seizures |
| ≤ 2 Minutes |
23 (46) |
17 (74) |
1 (4) |
5 (22) |
0.55 |
0.76 |
0.24 |
| >2 Minutes |
27 (54) |
17 (63) |
4 (15) |
6 (22) |
0.7 |
0.3 |
| Postictal period |
| ≤ 2 Minutes |
25 (50) |
20 (80) |
1 (4) |
4 (16) |
0.16 |
0.82 |
0.18 |
| >2 Minutes |
25 (50) |
14 (56) |
4 (16) |
7 (28) |
0.64 |
0.36 |
| Drug responsiveness |
| Yes |
40 (80) |
25 (62.5) |
5 (12.5) |
10 (25) |
0.34 |
0.69 |
0.31 |
| No |
10 (20) |
9 (90) |
0 (0) |
1 (10) |
0.9 |
0.1 |
Table 8: Association of SCN1A Thr1067Ala polymorphism genotypes with clinical features of Juvenile myoclonic epilepsy patients.
| Variables |
LGS patients (%) |
AA |
AG |
GG |
p value |
A allele frequency |
G allele frequency |
| Onset of seizures |
| ≤1 year of Age |
26 (52) |
18 (69) |
6 (23) |
2 (8) |
0.007 |
0.81 |
0.19 |
| >1 year of Age |
24 (48) |
24 (100) |
0 (0) |
0 (0) |
1 |
0 |
| Frequency of seizures |
| ≤3 per Day |
28 (56) |
24 (86) |
2 (7) |
2 (7) |
0.35 |
0.89 |
0.11 |
| >3 per Day |
22 (44) |
18 (82) |
4 (18) |
0 (0) |
0.91 |
0.09 |
| Duration of seizures |
| ≤5 Seconds |
24 (48) |
18 (75) |
4 (17) |
2 (8) |
0.19 |
0.83 |
0.17 |
| >5 Seconds |
26 (52) |
24 (92) |
2 (8) |
0 (0) |
0.96 |
0.04 |
| Drug responsiveness |
| Yes |
42 (84) |
36 (86) |
4 (9.5) |
2 (4.5) |
0.48 |
0.9 |
0.1 |
| No |
8 (16) |
6 (75) |
2 (25) |
0 (0) |
0.88 |
0.12 |
Table 9: Association of SCN1A Thr1067Ala polymorphism genotypes with clinical features of Lennox-Gastaut syndrome patients.
| Variables |
JME patients (%) |
GG |
GA |
AA |
p-value |
G allele frequency |
A allele frequency |
| Onset of seizures |
| ≤15 years of Age |
34 (68) |
28 (82) |
4 (12) |
2 (6) |
0.46 |
0.88 |
0.12 |
| >15 years of Age |
16 (32) |
15 (94) |
0 (0) |
1 (6) |
0.94 |
0.06 |
| Frequency of seizures |
| ≤1 per month |
21 (42) |
17 (81) |
3 (14) |
1 (5) |
0.47 |
0.88 |
0.12 |
| >1 per month |
29 (58) |
26 (90) |
1 (3) |
2 (7) |
0.91 |
0.09 |
| Duration of seizures |
| ≤2 Minutes |
23 (46) |
20 (87) |
1 (4) |
2 (9) |
0.59 |
0.89 |
0.11 |
| >2 Minutes |
27 (54) |
23 (85) |
3 (11) |
1 (4) |
0.91 |
0.09 |
| Postictal period |
| ≤2 Minutes |
25 (50) |
22 (88) |
1 (4) |
2 (8) |
0.7 |
0.9 |
0.1 |
| >2 Minutes |
25 (50) |
21 (84) |
3 (12) |
1 (4) |
0.9 |
0.1 |
| Drug Responsiveness |
| Yes |
40 (80) |
34 (85) |
4 (10) |
2 (5) |
0.42 |
0.9 |
0.1 |
| No |
10 (20) |
9 (90) |
0 (0) |
1 (10) |
0.9 |
0.1 |
Table 10: Association of SCN2A Arg19Lys polymorphism genotypes with clinical features of Juvenile myoclonic epilepsy patients.
| Variables |
LGS patients (%) |
GG |
GA |
AA |
p value |
G allele frequency |
A allele frequency |
| Onset of seizures |
| ≤1 year of Age |
26 (52) |
20 (77) |
4 (16) |
2 (7) |
0.43 |
0.85 |
0.15 |
| >1 year of Age |
24 (48) |
22 (92) |
2 (8) |
0 (0) |
0.96 |
0.04 |
| Frequency of seizures |
| ≤3 per Day |
28 (56) |
22 (79) |
4 (14) |
2 (7) |
0.59 |
0.86 |
0.14 |
| >3 per Day |
22 (44) |
20 (91) |
2 (9) |
0 (0) |
0.95 |
0.05 |
| Duration of seizures |
| ≤5 Seconds |
24 (48) |
20 (83) |
2 (8.5) |
2 (8.5) |
0.43 |
0.88 |
0.12 |
| >5 Seconds |
26 (52) |
22 (85) |
4 (15) |
0 (0) |
0.92 |
0.08 |
| Drug responsiveness |
| Yes |
42 (84) |
36 (86) |
4 (9) |
2 (5) |
0.48 |
0.9 |
0.1 |
| No |
8 (16) |
6 (75) |
2 (25) |
0 (0) |
0.88 |
0.12 |
Table 11: Association of SCN2A Arg19Lys polymorphism genotypes with clinical features of Lennox-Gastaut syndrome patients.
Clinical features of JME patients and their association with SCN1A Thr1067Ala polymorphism genotypes
No significant difference in SCN1A Thr1067Ala polymorphism genotypic distribution was found in JME patients with respect to any of the clinical features as shown in Table 8.
Clinical features of LGS patients and their association with SCN1A Thr1067Ala polymorphism genotypes
A significant difference in SCN1A Thr1067Ala polymorphism genotypic distribution was found in LGS patients with respect to age at onset of seizures with a p value of 0.007 as shown in Table 9. The mutant G allele of this polymorphism may be implicated in the early onset of seizures in LGS patients.
Clinical features of JME patients and their association with Arg19Lys polymorphism genotypes
No significant difference in SCN2A Arg19Lys polymorphism genotypic distribution was found in JME patients with respect to any of the clinical features as shown in Table 10.
Clinical features of LGS patients and their association with SCN2A Arg19Lys polymorphism genotypes
No significant difference in SCN2A Arg19Lys polymorphism genotypic distribution was found in LGS patients with respect to any of the clinical features as shown in Table 11.
Discussion
In the present study, the role of alterations in sodium channel genes SCN1A and SCN2A in two epilepsy syndromes i.e., JME and LGS was analysed. We studied two genetic polymorphisms, SCN1A Thr1067Ala and SCN2A Arg19Lys in these two epilepsy syndromes. Our observations indicate a negative association between the SCN1A Thr1067Ala polymorphism and occurrence of LGS. There was a significant difference in the genotypic distribution of this polymorphism between LGS patients and control subjects (p=0.008), and the percentage frequency of mutant homozygous (GG) plus heterozygous (AG) genotypes was lower in LGS. This correlated with the significantly lower mutant ‘G’ allele frequency observed in LGS patients (p=0.04), in comparison to healthy control subjects. Assessment of susceptibility/risk of LGS in association with inheritance of SCN1A Thr1067Ala polymorphism mutant GG genotype, revealed an odds ratio of 0.21 (95% CI 0.07 to 0.66, p=0.005) which is in sync with the differential genotypic/ allelic distribution in LGS. It appears from these findings that the mutant ‘G’ allele of SCN1A Thr1067Ala polymorphism in some way safeguards against the development of LGS, though it is premature to comment on the underlying pathophysiology. The protective role of this mutant ‘G’ allele in relation to LGS is a normal observation, which has not been reported earlier and needs further validation. A contradictory finding to this tenet which we noted in our LGS study group was a significant association of heterozygous and mutant homozygous genotypes of this polymorphism with an earlier age of onset of seizures i.e., less than one year (p=0.007).
The SCN1A Thr1067Ala results in change of amino acid threonine to alanine at a highly consensus conserved site in the coding region of the SCN1A, and possibly affects functioning of the inactivation gate in the cytosol regulating efflux and influx of sodium ions. Another possibility is that this polymorphism is in linkage disequilibrium with some other genetic variants of the same gene that affect risk of developing epilepsy.
Unlike our findings in LGS, we didn’t observe any significant difference in JME patients, either in the genotypic or mutant allele frequency distribution of SCN1A Thr1067Ala polymorphism, in comparison to healthy controls. The odds ratio determined as a measure of contribution of SCN1A Thr1067Ala polymorphism mutant homozygous and heterozygous genotypes, to the risk of JME, was also found to be non-significant (p=0.37 and 0.45 respectively).
A study by Khan et al. showed that SCN1A Thr1067Ala polymorphism may play an important role in development of various types of epilepsy viz., idiopathic, symptomatic and generalized [11]. SCN1A variant R604H in exon 11 was found in two probands from unrelated families with JME [12]. Three SNPs (R542Q, G1081R and T1174S) of SCN1A were found in sporadic cases of epilepsy although the family records were not available [37]. Of these 3 SNPs, R542Q and T1174S were observed in JME [38]. Only a minority of LGS patients have been shown to have a SCN1A pathogenic variant that is associated with an abnormal phenotype or increased disease risk [23].
In our study, results for SCN2A Arg19Lys polymorphism association with epilepsy syndromes were different from those of earlier studies. We observed a significant association of SCN2A Arg19Lys polymorphism with LGS (p=0.003), with the frequency of mutant homozygous (AA) plus heterozygous (GA) genotypes being higher in LGS patients than in control subjects. An increased risk of LGS was also observed in relation to heterozygous GA genotype of this polymorphism with an odds ratio of 4.24. However, no association of SCN2A Arg19Lys polymorphism with JME was observed in our study.
SCN2A Arg19Lys polymorphism was studied in German idiopathic generalized epilepsy patients and failed to show any association of this polymorphism with epilepsy susceptibility [39]. Nakayama et al. carried out the similar study in Japanese population and also did not find any association of this polymorphism with epilepsy [40]. A study done in Taiwan similarly failed to show any association of Arg19Lys polymorphism in epilepsy patients [41]. A study of Arg19Lys polymorphism in Indian population also showed no association with susceptibility to idiopathic, symptomatic and generalized epilepsy [21].
There are several limitations of the present study: the sample size was low and only two SNPs in sodium channel genes were screened in the study population. However, the role and presence of other genetic variations in the same gene or in other candidate genes cannot be ignored. Also, an in vitro functional study to examine the electrophysiological effects of the SNPs and mutations may provide insight into the molecular mechanism(s) underlying the pathogenesis of epilepsy syndromes. In summary, we observed a differential role of genetic variations in the SCN1A and SCN2A genes coding for alpha chains of sodium channels in JME & LGS epilepsy phenotypes. It would be worthwhile to study these polymorphisms at the functional level and also to replicate them in larger cohorts.
Conclusion
Both SCN1A Thr1067Ala and SCN2A Arg19Lys polymorphisms appear to influence the development of LGS, though in reciprocal directions. Inheritance of mutant allele of SCN1A Thr1067Ala polymorphism may have a dampening effect on Na+ channel firing and neuronal excitability, thereby leading to elevation of threshold for generation of seizures and other phenotypic effects in LGS. The mutant allele of SCN2A Arg19Lys polymorphism on the contrary, has a likely positive stimulatory role in action potential generation and thus increases the susceptibility to LGS. Neither SCN1A Thr1067Ala nor SCN2A Arg19Lys polymorphisms seem to modulate the pathophysiology or risk of development of JME.
These observations need to be substantiated further by more elaborate investigations across wide sections of different populations. Dissection of the molecular pathogenetic effects of these Na+ channel gene alterations would pave the way for a better understanding of epilepsy syndromes and more rational therapeutic drug design.
Acknowledgement
We would like to thank Indian council of medical research (ICMR), New Delhi for providing financial assistance for this research.
Conflict of Interest
None
21689
References
- Pringsheim T, Fiest K, Jette N (2014) The international incidence and prevalence of neurologic conditions: How common are they? Neurol 83: 1661-1664.
- Beghi M, Beghi E, Cornaggia CM, Gobbi G (2006) Idiopathic generalized epilepsies of adolescence. Epilepsia 47: 107-110.
- Delgado-Escueta AV (2007) Advances in genetics of juvenile myoclonic epilepsies. Epilepsy Curr 7: 61-67.
- Allen AS, Berkovic, Cossette P, Delanty N, Dlugos D, et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501: 217-221.
- Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF (2015) Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol 88: 203-217.
- Bertrand D (2002) Neuronal nicotinic acetylcholine receptors and epilepsy. Epilepsy Curr 2: 191-193.
- Kohling R (2002) Voltage-gated sodium channels in epilepsy. Epilepsia 43: 1278-1295.
- Rossi MA (2013) Juvenile myoclonic epilepsy: When will it end. Epilepsy Curr 13: 148-149.
- Auvin S(2007) Treatment of myoclonic seizures in patients with juvenile myoclonic epilepsy. Neuropsychiatr Dis Treat 3: 729-734.
- Ma S, Blair MA, Abou-Khalil B, Lagrange AH, Gurnett CA, et al. (2006) Mutations in the GABRA1 and EFHC1 genes are rare in familial juvenile myoclonic epilepsy. Epilepsy Res 71:129-134.
- Zifkin B, Andermann E, Andermann F (2005) Mechanisms, genetics, and pathogenesis of juvenile myoclonic epilepsy. Curr Opin Neurol 18: 147-153.
- Mastrangelo M, Lennox-Gastaut (2017) Syndrome: A state of the art review. Neuropediatrics 48: 143-151.
- Asadi-Pooya AA, Gastaut L (2017) Syndrome: A comprehensive review. Neurological sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology.
- Rijckevorsel K (2008) Treatment of Lennox-Gastaut syndrome: Overview and recent findings. Neuropsychiatr Dis Treat 4: 1001-1019.
- Shen H, Zhou Q, Pan X, Li Z, Wu X, et al. (2017) Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355.
- Catterall WA, Noebels JL, Avoli ML, Rogawski MA, Olsen RW (2012) Sodium channel mutations and epilepsy. In: The Jasper's Basic Mechanisms of the Epilepsies, Bethesda, USA.
- Marban E, Yamagishi, Tomaselli GF (1998) Structure and function of voltage-gated sodium channels. J Physiol 508: 647-657.
- de Lera Ruiz M, Kraus RL (2015) Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem 58: 7093-7118.
- Yu FH, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biol 4: 207.
- Catterall WA (2014) Structure and function of voltage-gated sodium channels at atomic resolution. Exp Physiol 99: 35-51.
- Lakhan R, Kumari R, Misra UK, Kalita J, Pradhan S, et al. (2009) Differential role of sodium channels SCN1A and SCN2A gene polymorphisms with epilepsy and multiple drug resistance in the north Indian population. Br J Clin Pharmacol 68: 214-220.
- Raymond CK, Castle J, Garrett-Engele P, Armour, Kan Z, et al. (2004) Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. J Biol Chem 279: 46234-46241.
- Miller IO, Sotero de Menezes MA (1993) SCN1A-related seizure disorders.
- Krauss G, Baranano K (2014) “Idiopathic" and "genetic" generalized epilepsies intersect. Epilepsy Curr 14: 81-83.
- Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G (2017) Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140: 1316-1336.
- Carroll LS, Woolf R, Ibrahim Y, Williams HY, Dwyer YS (2016) Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation. Psychiatr Genet 26: 60-65.
- Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki (2009) De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology 73: 1046-1053.
- Alekov A, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H (2000) A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J Physiol 529: 533-539.
- Vilin YY, Ruben PC (2001) Slow inactivation in voltage-gated sodium channels: Molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35: 171-190.
- Lombardo AJ, Kuzniecky R, Powers RE, Brown GB (1996) Altered brain sodium channel transcript levels in human epilepsy. Mol Brain Res 35: 84-90.
- Meisler MH, Kearney JA (2005) Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115: 2010-2017.
- Ito M, Shirasaka Y, Hirose S, Sugawara T, Yamakawa K (2004) Seizure phenotypes of a family with missense mutations in SCN2A. Pediatr Neurol 31: 150-152.
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld S (2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24: 343-345.
- Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima Y (2003) Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126: 531-546.
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma (2001) A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proceedings of the National Academy of Sciences of the United States of America 98: 6384-6389.
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T (2001) A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus--and prevalence of variants in patients with epilepsy. Am J Hum Genet 68: 866-873.
- Singh R, Andermann E, Whitehouse WP, Harvey AS, Keene DL (2001) Severe myoclonic epilepsy of infancy: Extended spectrum of GEFS+? Epilepsia 42: 837-844.
- Haug K, Hallmann K, Rebstock J, Dullinger J, Muth S, et al. (2001) The voltage-gated sodium channel gene SCN2A and idiopathic generalized epilepsy. Epilepsy Res 47: 243-246.
- Nakayama, Yamamoto N, Hamano K, Iwasaki N, Ohta M (2002) Failure to find evidence for association between voltage-gated sodium channel gene SCN2A variants and febrile seizures in humans. Neurosci Lett 329: 249-251.
- Chou IC, Tsai FJ, Huang CC, Shi YR, Tsai CH (2003) The lack of association between febrile convulsions and polymorphisms in SCN1A. Epilepsy Res 54:53-57.

