Keywords
In-vitro fertilization; Oocyte; Sperm; Embryo; Endometrium and Methods of evaluation
Introduction
In-vitro Fertilization (IVF) is one of the methods for assisted conception that became popular after the birth of Louise Brown in 1978. This method differs from Intra Uterine Insemination (IUI) in which the sperm is deposited in the uterus and allowed to fertilize the egg in-vivo. In IVF, the egg and the sperm are fertilized outside the body in a tissue culture dish. The eggs are collected trans-vaginally using an ultrasound after multiple follicles are induced to develop by gonadotropins. They eggs retrieved may be fertilized through conventional IVF, Intracytoplasmic Sperm Injection (ICSI) or Intracytoplasmic Morphologically Selected Sperm Injection (IMSI). ICSI or IMSI is applied when the sperm count is low [1].
Once fertilization occurs, the zygotes are allowed to develop for 3 or 5 days in the incubator before they are transferred to the uterus. Where there are 5 embryos or more that are 8 cells on a day 3 and without fragments, they may be allowed to proceed to day 5. Day 5 or 6 embryos are expected to become blastocysts, however some embryos that are 8 cells on day 3 may fail to reach the blastocyst stage on day 5. This may allow for the selection of potentially viable ones based on the rate of development [2], in this case the ones that made it to the blastocyst stage. Whether embryos are selected on day 3 or 5, they are transferred into the uterus through a catheter under ultrasound guidance. If the embryo is viable, it may attach to the endometrium and pregnancy may results.
Initially, clinicians and patients preferred more than one embryo to be transferred. This is because if three embryos are transferred, one, two, three or none of them may implant. In the UK for example between 1992 and 2006, more babies were born through IVF when more than one embryo where transferred (Figure 1). Between 2010 and 2011 (Figure 2), the rate slightly dropped as the practice of multiple embryo transfer decline. Multiple embryo transfer is being discouraged because of the risk of multiple births. Multiple births pose significant risk to the mother and the health and wellbeing of babies (Table 1). Because of this, the Human Fertilization and Embryology Authority (HFEA) introduced a multiple births policy in 2009 which recommends only one embryo to be transferred even if more are available, this has resulted in a drop of multiple live birth rate from around 24% to around 17% in 2012 [6].
| Risks |
Percentage |
| |
Singleton |
Multiple |
| Pregnancy induced hypertension |
1-5% |
20% |
| Pre-eclampsia |
2-10% |
30% |
| Gestational diabetes |
4% |
12% |
| Maternal mortality |
0.00% |
0 |
Table 1: Comparison of singleton and multiple pregnancy risks to the mother. (Adapted from [5]).
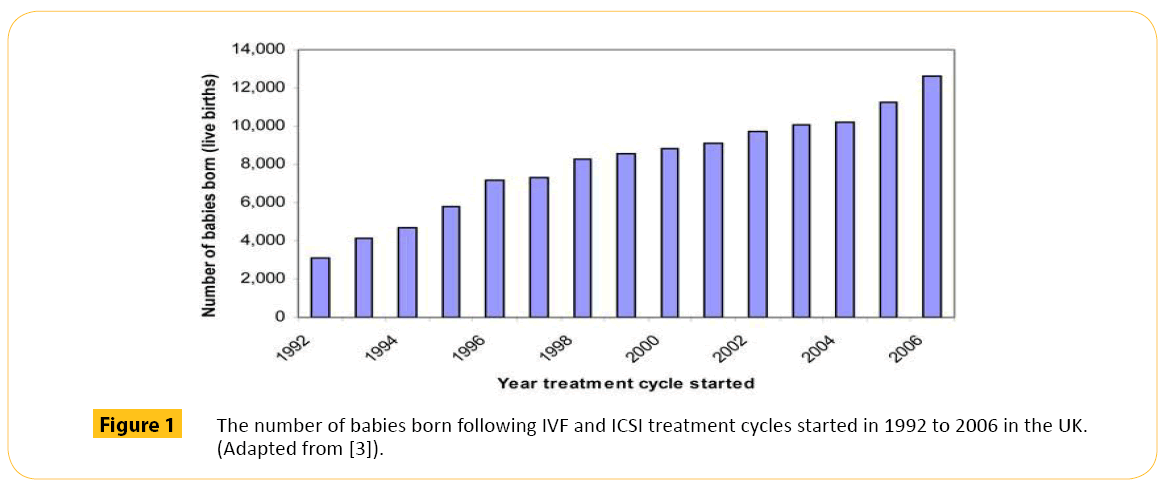
Figure 1: The number of babies born following IVF and ICSI treatment cycles started in 1992 to 2006 in the UK. (Adapted from [3]).
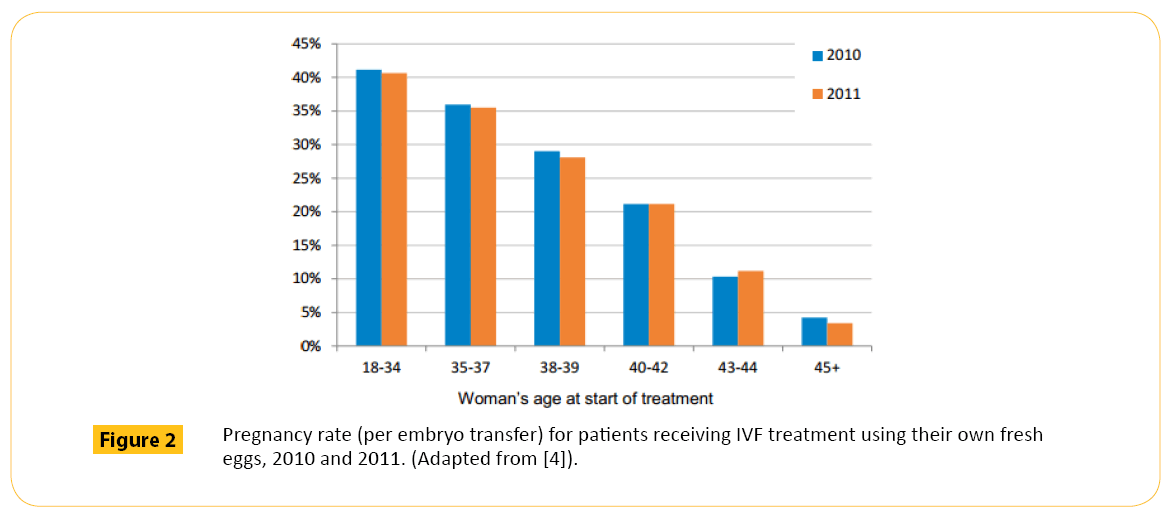
Figure 2: Pregnancy rate (per embryo transfer) for patients receiving IVF treatment using their own fresh eggs, 2010 and 2011. (Adapted from [4]).
Though this policy was effective towards reducing multiple births, it caused a slight decrease in the success rate for live birth recorded across the fertility clinics. This is not surprising because the single embryo for transfer is usually selected by morphological assessment. Morphological assessment alone cannot tell when an embryo is unhealthy at the molecular level and if such an unhealthy embryo is transferred, it may not result in live birth.
Even when a viable embryo is transferred, the endometrium must be receptive before pregnancy can occur. It is therefore important that emphasis be made on the evaluation of the endometrium just as much is being done to improve on the evaluation of embryo selection [7].
The traditional method for evaluating the viability of an embryo is morphological assessment. This method is subjective because evaluations are based on the number of blastomeres in the embryo, symmetry of the blastomeres and degree of fragmentation [8,9]. Morphological assessment does not detect chromosomal abnormality or defects in critical cellular processes like protein synthesis, transcription and metabolism that can impact on viability of an embryo.
Presently, the only definitive method being used to evaluate if an embryos is normal or abnormal other than the morphological criteria is pre-implantation genetic screening (PGS). The method used in sampling for PGS is invasive. Though a biopsy free method of sampling DNA has been published by Palini et al. [10], research in this area are still preliminary [11]. In the early years of PGS, Samples were analyzed using fluorescence in-situ hybridization (FISH). The FISH technology has been criticized as inefficient [12]. A review by Braude [13] highlighted over 12 randomized trials showing either no benefit or adverse effect of cleavage-stage biopsy and FISH. It also showed that the performance of FISH in aneuploidy screening may not be related to the technology alone but also the complex interactions within the embryos. There are evidences that embryos are compartmentalized [14]. The implication of this is that the DNA of an abnormal cell within an embryo could be localized in one part of the embryo and absent in the others as suggested by Braude. Thus, it is the complexity of an embryo not necessarily the technology that has contributed to the failure of PGS as a method for selecting single viable embryo. Changing this technology to comparative genomic hybridization (CGH) or single-nucleotide polymorphism (SNP) arrays is no more valuable as an alternative method that will encourage single embryo transfer [13]. This is because CGH or SNP technologies will not address the issue of compartmentalization in embryos, a contributory factor to it complexity.
Significant progress has been made towards improving knowledge about the genetic sequence of an embryo. However, the phenotype the embryo will manifest from this sequence may be altered by carrier molecules responsible for transferring this information. PGS alone cannot tell when this happened. The application of technologies to capture all that is going on in the embryo at the genomic, proteomic, transcriptomic or metabolomic level is critical to generating vital information about the complex interactions within the embryo. These technologies are collectively called omics, and are being introduced into the embryology laboratories individually and used in conjunction with morphology method to assist in the selection of embryos [15].
Already, proteomic which provides information about the protein an embryo secretes have demonstrated potentials to distinguish between culture media from implanting and non-implanting embryo [16]. The advantage of this approach is that it is noninvasive. The transcriptomics profile of embryos have been interrogated to tell when and where gene expression is turned on or off as well as the number of RNAs involved to know how the gene is being expressed [17]. This can be a powerful means of assessing embryos that are viable from those that are not in the future. There are also growing evidence that metabolic profiles of spent embryo culture media differ between embryos that implant and those that fail to implant and may be used to predict reproductive potential [18-21]. Technologies that assess omic profiles are available, the problem is that they are commonly applied to assess individual pathway. Any single technology that is able to assess the pathways of genomic, proteomic, transcriptomic and metabolomic from a drop of spent media may be the greatest innovation the IVF world is waiting for. This is because it will be able to tell if the embryo has normal genes, proteins, ribonucleic acids and metabolic products. Any single embryo that is considered normal by this technology will be capable of producing a clinical pregnancy. If a single technology is able to assess all these parameters, the possibilities of selecting a single embryo from a cohort that is viable may be increased. This may discourage multiple embryo transfer and reduce multiple births. A technology like this should be simple, rapid and affordable and should only be introduced after it has been properly validated.
Morphological assessment of embryo
Traditionally, the quality of gametes and embryos developed in IVF are assessed based on their morphology. This morphological evaluation allows embryologists to determine the level of development of the embryo and to select potential viable ones for transfer [8]. There are different morphological grading schemes currently being used to monitor embryo developmental potential. Though this method of assessing viability is being practiced worldwide, it is very difficult to validate outcome from morphology assessment across centers because of the variation in embryo grading schemes adopted by different fertility clinics.
While national consensus schemes exist in some countries, many other countries are yet to copy this model making it difficult for validation on a worldwide scale. Attempts have been made in this direction, an example is the Istanbul panel invited to develop an international consensus on embryo assessment [22]. It was agreed by this panel that the first assessment of an embryo development should be done on the zygote, after insemination or ICSI. The fertilized egg is assessed for the appearance of pronuclei (a pronucleus is the nucleus of a sperm or an egg cell). The appearance of two pronucle is the first sign of successful fertilization. When pronuclei are observed, their number and size as well as the number and symmetry of the nucleolar precursor bodies (Table 2) should be reported to help guide the selection of viable embryos.
| Grade |
Rating |
Description |
| 1 |
Symmetrical |
Equivalentto Z1and Z2 |
| 2 |
Non symmetrical |
Other arrangements,including peripherallysited pronuclei |
| 3 |
Abnormal |
Pro nuclei with 0or 1NPB |
Table 2: Consensus Fertilization stage grading system. (Adapted from [22]).
When the embryo begins to cleave, the rate of division, symmetry of the blastomeres, multi-nucleation and the degree of fragmentation (Table 3) in the developing embryos are assessed to tell the ones that are normal by the morphological criteria.
| Grade |
Rating |
Description |
| 1 |
Good |
<10% fragmentation, stage specific cell size,no multi nucleation |
| 2 |
Fair |
Upto 25% fragmentation,stage specific cellsize for majority of cells, no evidence ofmultinucleation |
| 3 |
Poor |
Severe fragmentation (>25%),cell size not stage specific, evidence of multinucleation |
Table 3: Consensus Cleavage stage embryo scoring system. (Adapted from [22]).
In the blastocyst stage embryos, development and viability is gauged by the degree of blastocoele expansion and hatching status as well as the size and compactness of the inner cell mass (Table 4). The cohesiveness and number of trophectoderm (TE) cells are also important consideration.
| Grade |
Rating |
Description |
| Stage of development |
|
Early |
| |
1 |
|
Blastocyst |
| 2 |
|
Expanded |
| 3 |
|
Hatched/Hatching |
| Inner Cell Mass |
1 |
Good |
Prominent, easily discernible, with many cells that are compacted and tightly adhered |
| |
2 |
Fair |
Easily discernible, with many cells loosely grouped together |
| |
3 |
Poor |
Difficult to discern with few cells |
| |
1 |
Good |
Many cell forming A cohesive epithelium |
| |
2 |
Fair |
Few cells forming a loose epithelium |
| |
3 |
Poor |
Very few cells |
Table 4: Consensus blastocyst scoring system. (Adapted from [22]).
The drive to improve on the predictive capacity of morphological assessment is largely due to its low cost and the relative ease to implement in IVF laboratories [23]. Despite these advantages, the subjective nature of this method is a cause for concern even with adequate expertise [24,25]. This is the reason why embryos classified as low grade often produce viable pregnancy. Efforts to find an alternative method of embryo assessment that will provide more detailed information about the embryo viability have led to the development of a dynamic process which captures several critical stages in between fertilization and blastocyst formation that were not observed when embryos were assessed on specific time by morphological assessment alone. This dynamic process is called time lapse monitoring
Time lapse monitoring
Time-lapse monitoring is capable of allowing continuous observation and analysis of cellular dynamics over long periods of time in an embryo. Embryos can be continuously monitored during their developmental stage in an embryoscope (Figure 3) which is a microscope with a chamber containing the embryo under optimum culture conditions

Figure 3: Photographs taken using an embryoscope showing different grading for the same embryo at different time points during a time frame of 6 hours. (Adapted from [26]).
Proponents of this technology argue that the poor sensitivity of the current morphological assessment method, which only classifies embryo based on observations at a specific time to avoid over exposure of embryos outside the incubator, have been addressed with time lapse technology [23]. It is believed that with this technology, dynamic markers of embryo viability will differentiate between implanting and non- implanting embryos.
Presently, the parameters used in time lapse monitoring for blastocyst prediction include: (i) first cytokinesis, (ii) 2-cell stage and (iii) 3-cell stage [27]. These parameters have been shown to be incapable of predicting pregnancy [28]. The precision of this method of selection may be affected by blastomere overlapping [29] which may mislead the embryologist because this system does not permit rotation that is normally used to separate the blastomeres during morphological evaluation under the microscope. Also, the current parameters used to predict embryo potential in Time lapse captures until 5 cell stage (Figure 4) where embryo development is majorly directed by the maternal genome because the embryo has not yet acquire the capacity to direct its own development [27]. Prediction based on this model may not represent the true embryo status after the embryo activates its own genome. An embryo can have a good morphology and time-lapse grading and still not produce pregnancy because morphological evaluation alone cannot tell of other defects in the embryo [31]. This drawback is the driving force for an improved technology that could provide the picture of an embryo at the molecular level. One of these molecular innovations employed to address this need is called pre-implantation genetic screening (PGS)
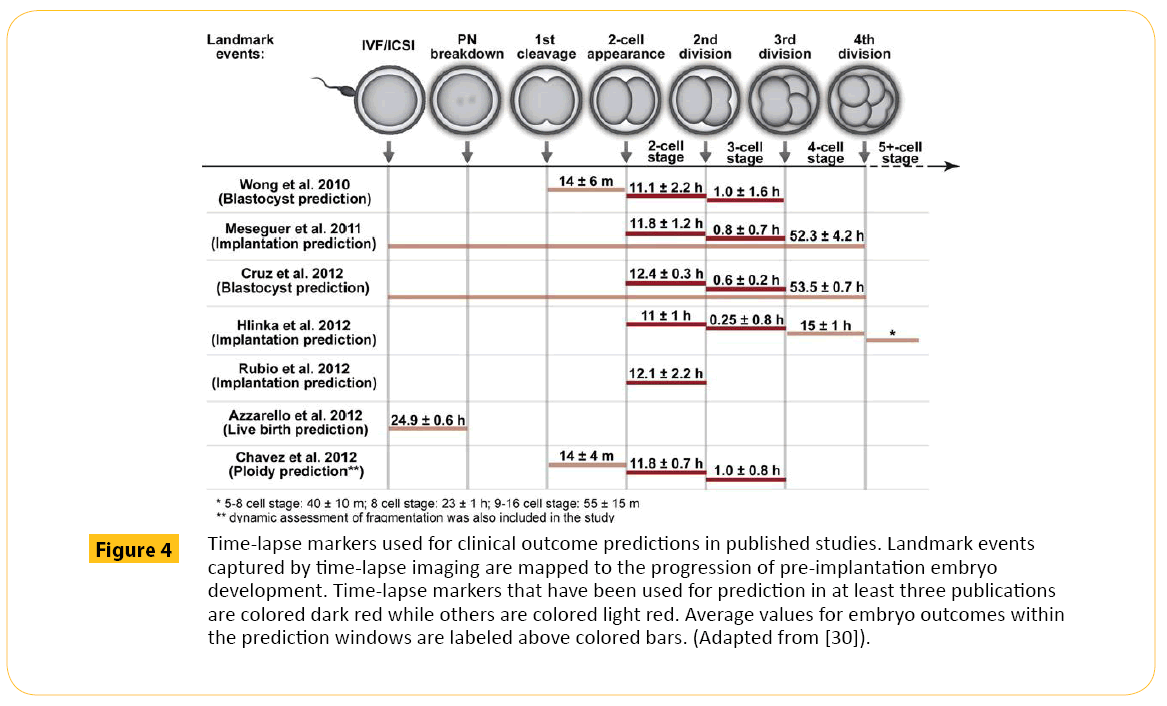
Figure 4: Time-lapse markers used for clinical outcome predictions in published studies. Landmark events captured by time-lapse imaging are mapped to the progression of pre-implantation embryo development. Time-lapse markers that have been used for prediction in at least three publications are colored dark red while others are colored light red. Average values for embryo outcomes within the prediction windows are labeled above colored bars. (Adapted from [30]).
Pre-implantation genetic screening: Invasive technologies
Embryos produced through IVF or ICSI may have normal morphological grading and still have genetic defects or poor morphological grade with normal genetic profile. This is because morphology alone does not indicate genetic defect in an embryo. In IVF, genetic defects in an embryo can be detected by preimplantation genetic screening. These screening are necessary to identify embryos affected with chromosomal defects to prevent them from being transferred. The exclusion of these embryos from the cohort for transfer is important because they are less likely to implant, may result in miscarriage or the birth of genetically abnormal child [32].
Over 50% of IVF embryos have abnormal chromosomes increasing to up to 80% in women over 40 years of age [33]. Despite the presence of these abnormalities, few embryos arrest between day 3 and 5. Most progressed into the blastocyst stage [34]. The implication of this is that genetically incompetent embryos that are likely going to miscarriage or failed to implant cannot be detected by the rate of embryo development alone observed morphologically. This has placed PGS as a better method to differentiate between viable and non-viable embryos than the current morphological technique.
PGS cases were initially done with Fluorescence in situ hybridization (FISH). Contrary to expectation, results from numerous studies did not show that the PGS-FISH was capable of improving live birth rates in patients of advanced maternal age, with recurrent implantation failure or repeated pregnancy loss [35-46]. This is because FISH screening does not make it possible to test for all 23 chromosome pairs. At best only 12 of the 23 chromosome pairs of an embryo can be analyzed using FISH thereby allowing some chromosomal abnormalities to be missed [33]. This limitation in FISH technology indicates a need for newer technologies like metaphase comparative genomic hybridization (mCGH), array Comparative Genomic Hybridization (aCGH), Qualitative Polymerase Chain Reaction(qPCR), Single Nucleotide Polymorphism (SNP) array and more recently Next- Generation Sequencing (NGS). These technologies are capable of analyzing all the 23 chromosome pairs from a single cell and have been shown to improve IVF clinical pregnancy rates by 32% in a randomized controlled trial [47-54].
The problem with mCGH analysis is that it is more manual intensive and as such takes time to analyze the chromosomes of an embryo. Using this technology makes it difficult to select embryo for fresh transfer [54]. It is possible to have fresh transfer using aCGH because the process has been automated. The problem is that the presence of inversions and translocations may be missed by this approach [55]. SNP can detect not only copy number changes, but also inversions and translocations in chromosomes. The problem with SNP arrays are that it requires a lot of labor to analyze data and specialized protocol for detecting chromosome copy number variation (CNV) when whole-genome amplification from single cells are being performed [56]. NGS has the advantage of simultaneously screening for aneuploidy and single gene disorders in less time than the other techniques from a single cell and is also cost effective. One major limitation of NGS is the interpretation of the massive sequence data generated by the technology. Also, the accuracy of NGS needs further evaluation and validation before it should be implemented in the clinical selection of embryos [57].
Despite this advance in technologies for PGS, none of the tests is 100% reliable [56]. The initial setback in PGS-FISH was thought to be due to the FISH, however Braude [13] reported an investigation by Northrop which revealed that the setback in PGS-FISH may not be due to the FISH technology alone but also of the embryo complexity. This conclusion was arrived at when 58% of 50 cleavage stage embryos that were diagnosed as abnormal on cleavage-stage biopsy and FISH were found to be normal upon re-testing with microarray at the blastocyst stage. Also, more than half of the 21 embryos confirmed at the blastocyst stage to be aneuploid revealed inconsistency with the biopsy result of the cleavage stage embryos, while 12 of the embryos showed an abnormality different from that originally diagnosed. Braude [13] suggests that the embryos had different population of cells with different genetic compositions. He substantiates his argument with the result of blastocyst biopsied on three different locations on the trophectoderm and one in the inner cell mass (ICM). The result showed that the abnormality found at earlier biopsy was inconsistent with the one of the four locations biopsied. This means that the DNA of an abnormal cell within the cleavage-stage embryos was not uniformly distributed within the embryo. This compartmentalization of embryo has made diagnosis from any one area of the embryo unreliable because an embryo diagnosed as abnormal based on sample from one location may turn out normal if sample is collected from another location. From this finding, the argument that blastocyst stage biopsy is more representative than cleavage stage biopsy is questionable. Thus, it is the complexity in an embryo not necessarily the technology alone that has contributed to the failure of PGS as a method for selecting viable embryos. To tackle the complexity of an embryo holistically, it is important to understand its physiology.
Embryo physiology
After fertilization, a zygote is formed. The zygote is a product of the male and female chromatin evident by 2 pronuclei (Figure 5a). The fusion of these pronuclei leads to the formation of a new nucleus containing all of the embryo's genetic information (46 chromosomes) in a single cell. The fertilized, one-cell zygote is thought to be transcriptionally silent. In order to cleave into embryo, it will require maternal mRNAs and proteins from the egg for initial development until the time when it embryonic genome will be activated [64]. It has also been suggested that an embryo that is developing optimally will have less damage in this genome, in the protein synthesized as well as the mRNA produced [65]. Because of this minimal damage, it is expected that less energy will be spent by the embryo to correct any of this defect so the metabolism in such an embryo should be quiescent rather than active.
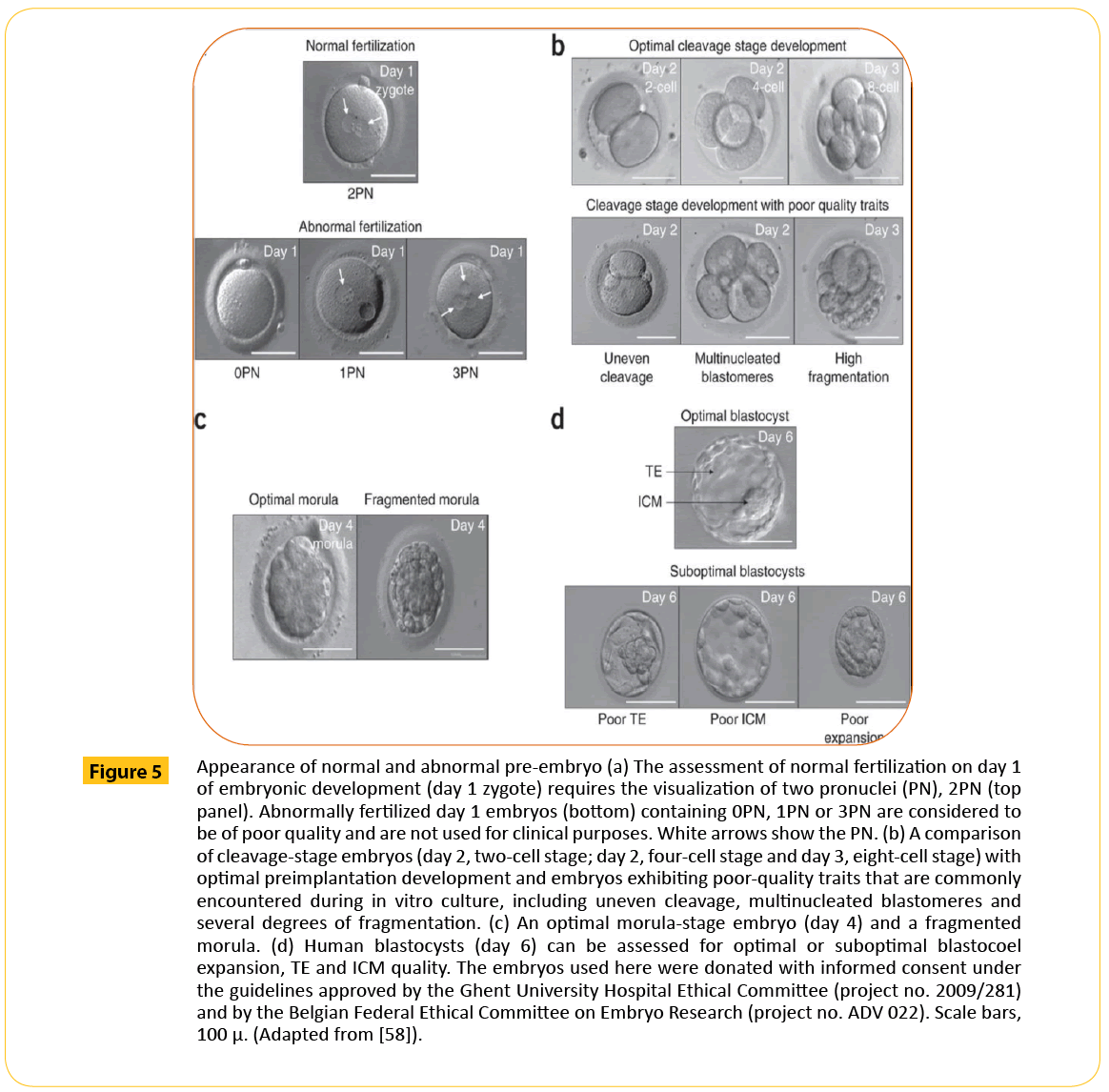
Figure 5: Appearance of normal and abnormal pre-embryo (a) The assessment of normal fertilization on day 1 of embryonic development (day 1 zygote) requires the visualization of two pronuclei (PN), 2PN (top panel). Abnormally fertilized day 1 embryos (bottom) containing 0PN, 1PN or 3PN are considered to be of poor quality and are not used for clinical purposes. White arrows show the PN. (b) A comparison of cleavage-stage embryos (day 2, two-cell stage; day 2, four-cell stage and day 3, eight-cell stage) with optimal preimplantation development and embryos exhibiting poor-quality traits that are commonly encountered during in vitro culture, including uneven cleavage, multinucleated blastomeres and several degrees of fragmentation. (c) An optimal morula-stage embryo (day 4) and a fragmented morula. (d) Human blastocysts (day 6) can be assessed for optimal or suboptimal blastocoel expansion, TE and ICM quality. The embryos used here were donated with informed consent under the guidelines approved by the Ghent University Hospital Ethical Committee (project no. 2009/281) and by the Belgian Federal Ethical Committee on Embryo Research (project no. ADV 022). Scale bars, 100 μ. (Adapted from [58]).
Braude and colleagues [66] investigated the time in which human embryonic genome is activated by studying protein synthesis linked to transcriptional activation. They showed that this occurred following the 4-cell stage at approximately 8 cells. After embryonic genome activation (EGA), the embryo begins to compact and the blastomeres become smaller. This continuous until a morula is formed (Figure 5c), further development leads to the formation of a blastocyst (Figure 5d). The blastocyst comprises the trophectoderm (TE) and the inner cell mass (ICM) (Figure 5d).
Once an embryo assumes responsibility for its own development following the activation of its transcription mechanism, a relationship is established at the gene, RNA and metabolic level. The health status of the embryo can therefore be checked by assessing these complex interactions instead of the activities at only one pathway because of the relationship that exists. This can be exemplified by a metabolically quiescent embryo which is thought to have a stable genome, a normal proteome and transcriptome. This means that a viable embryo should only be selected after the genome, proteome, transcriptome and metabolome are interrogated.
Omics
This is a short description for technologies involving Genomics which provides data about the genome (genes), Proteomics which provides data about the proteome (protein), metabolomics which provides data about the metabolome (metabolites) and transcriptomics which provides information about the transcriptome (mRNAs). They are being introduced into embryology laboratory as a technique to assess embryo viability.
Genomics
Since the completion of the Human Genome Project, opportunity to diagnose the genome using high throughput technologies was created. These technologies have made it possible to analyze an individual genomic makeup to provide information relating to genes that are turned on or off. This information may provide a more precise method for distinguishing normal embryos from abnormal ones.
Human embryonic genome has been successfully interrogated with PGS technologies which exploit the natural process of hybridization such as FISH, mCGH, aCGH, SNP array and NGS. Initial technologies could not deliver on the high expectation of PGS due to technical limitation on the number of chromosome to be analyzed and time factor relating to fresh embryo transfer. Newer technologies have overcome these challenges and have delivered in term of improved outcome (Table 5).
| Reference |
Patient group |
Control group |
Method |
Day of Biopsy |
Outcome |
Improved outcome |
| Brooke Hodes-Wertz et al. [59] |
RPL, two or more losses (287) |
Expected RPL, SART data |
24,a CGH |
D3,D5 |
6.9% RPL 55% FCA 63% D5 50% D3 |
Yes |
| Schoolcraft et al. [60] |
AMA>35 yrs(30) |
No test, morphology, (30) |
24,SNP microarray |
D5 |
60.8% FCA 0% spont abort |
Yes |
| Forman et al. [61] |
Good prognosis SET vs DET |
DET |
qPCR-CCS 4 hours |
D5 |
66% FCA No twins |
Yes |
| Yang et al. [62] |
<35 yrs, no miscarriage, 1st IVF cycle, SET, (48) |
No test, morphology |
24,array CGH |
D5 |
71% FCA 69% ongoing preg(<20 wk) (55) |
Yes |
| Rubio et al. [63] |
Different indication |
Fresh blastocyst transfer |
24,array CGH |
D3 |
47.2%to 59.0% |
Yes |
RPL: Recurrent Pregnancy Loss; AMA: Advanced Maternal Age; SET: Single Embryo Transfer; DET: Double Embryo Transfer; FCA: Fetal Cardiac Activity; SART: Society for Assisted Reproduction Technologies; qPCR: Qualitative Polymerase Chain Reaction; CCS: Comprehensive Chromosome Screening; SNP: Single Nucleotide Polymorphism; aCGH: Array Comparative Genomic Hybridization; D3: Day 3; D5: Day 5
Table 5 Improved outcome of IVF after 24 chromosome analysis.
Proteomic
The proteome is vital for cellular function and represents all the proteins translated from a cell's speci?c gene expression products. There are over 1 million of these proteins and their function can be affected by internal and external factors during the processes of translation, post-translational modi?cation and interactions [18]. This makes it important that the human embryonic proteome is fully understood so that any alteration could be detected.
It is thought that a viable embryo possesses a unique protein secretome which is secreted into the surrounding culture medium. Consequently, a non-invasive proteomic analysis of the secretome of human embryos throughout pre-implantation development may assist in revealing secreted factors that reflect developmental competence and viability. Initial analysis on the embryonic proteome was done using two-dimensional (2D) gel electrophoresis. Also, Western blotting has been used to identify the expression of known proteins or to detect posttranslational modifications, like phosphorylation, in relation to embryo development. Today, it is possible to identify groups of proteins within limited amounts of complex biological fluids and tissues using mass spectrometry (MS) and Protein microarrays [67].
Dominguez et al., [16] analyzed conditioned media from implanted and non- implanted embryo before transfer. There data showed that proteins like CXCL13 (BCL, B lymphocyte chemo-attractant), stem cell factor, TRAILR3, MIP-1b, and MSP-a where decreased in the blastocyst culture. They decreased in the expression of these factors indicates that these proteins have been consumed by the blastocysts. While soluble tumor necrosis factor receptor 1 and interleukin (IL)-10 increased significantly in media when a blastocyst was present. Interestingly, when comparing conditioned media from implanted vs. nonimplanted, two proteins, CXCL13 and granulocyte macrophage colony-stimulating factor, were found to have been consumed in conditioned media of implanted blastocysts. The CXCL13 is the same protein that decreased in the presence of blastocyst. In another follow-up research by Dominguez et al. [68], they analyzed protein secretome from an endometrial epithelial cell (EEC) coculture system with those from a sequential microdrop culture media system. Proteins like IL-6, PLGF, and BCL (CXCL13) in the EEC coculture system, showed increased expression with IL-6 being the most expressed protein. IL-6 was further probed in the spent media using ELISA assay, implanting embryos showed increase expression indicating that IL-6 may be a vital protein common in viable embryos. In contrast, FGF-4, IL- 12p40, VEGF, and uPAR, showed decreased expression.
Despite the huge efforts made, much still needs to be done to overcome the effects of limited template, low protein concentration, poor platform sensitivity, and limited protein database information. Other limitations include overwhelming presence of albumin, immunoglobulin, and other serum proteins in the culture media, making it difficult to identify the low expressed secreted embryonic proteins. Chromatographic approaches to remove such abundant proteins do exist and in combination with multidimensional fractionation will allow for the detection of proteins secreted by the embryo [67].
Transcriptomics
A transcriptome is a collection of RNAs derived from the DNA of a specific cell. The transcriptome includes ribosomal RNA (rRNA), messenger RNA (mRNA), transfer RNA (tRNA), micro RNA (miRNA), and other non-coding RNA (ncRNA) [69]. The genome is made up of deoxyribonucleic acid (DNA), a long, helical molecule that contains the information necessary for the sperm and egg to develop into a viable embryo. For this information to be useful, the DNA must be copied out into the corresponding molecules of ribonucleic acid (RNA). Studying the sequence of the RNA is therefore vital to the understanding of the sequence of the DNA from which it was derived from. This mean that when transcriptome is analyzed, it is possible to assess when and where gene expression is turned on or off as well as the number of RNAs involved to know how the gene is being expressed [18].
After the fusion of the sperm and egg, the resulting zygote is dependent on the information inherited from the maternal egg to direct its development. This is because the embryonic genes are still turned off (transcriptionally silent). In human, the embryonic gene becomes active at 4-8 cell stage which is maintained throughout adult life. Dobson et al., [70] reported several studies that documented key events that follow fertilization in humans, such as a decrease in abundance of individual mRNAs, activation of bulk mRNA transcription following a quiescent period and nascent expression of a few individual known genes that were previously characterized in other organisms. They also validated reports that demonstrate robust patterns of stage specific gene expression, representing maternal genes that are deactivated (down- regulated) and zygotic genes that are activated (upregulated) using their microarray studies.
DNA microarrays have been commonly used to study transcriptome of oocytes and embryos by measuring the relative amount of mRNA molecules that are present in a sample simultaneously for thousands of mRNA sequences (transcriptome), enabling comparison of the expression of thousands of genes in a given oocyte or embryo (Figure 6). Next generation sequencing technologies has brought light in this search through whole transcriptome shotgun sequencing (RNA seq) [72]. The advent of RNA seq has made genome-wide assessment of mRNA expression more reliable making it possible to indentify genes and groups of genes that are differentially expressed under various conditions, detection of co-expressed genes, and inference of the regulatory effect of genes among each other. Despite these technologies deployed to perform mRNA profiling in attempts to determine the factors that are responsible for embryo viability, no clear set of key genes has yet emerged, that can predict viable embryos from nonviable ones.
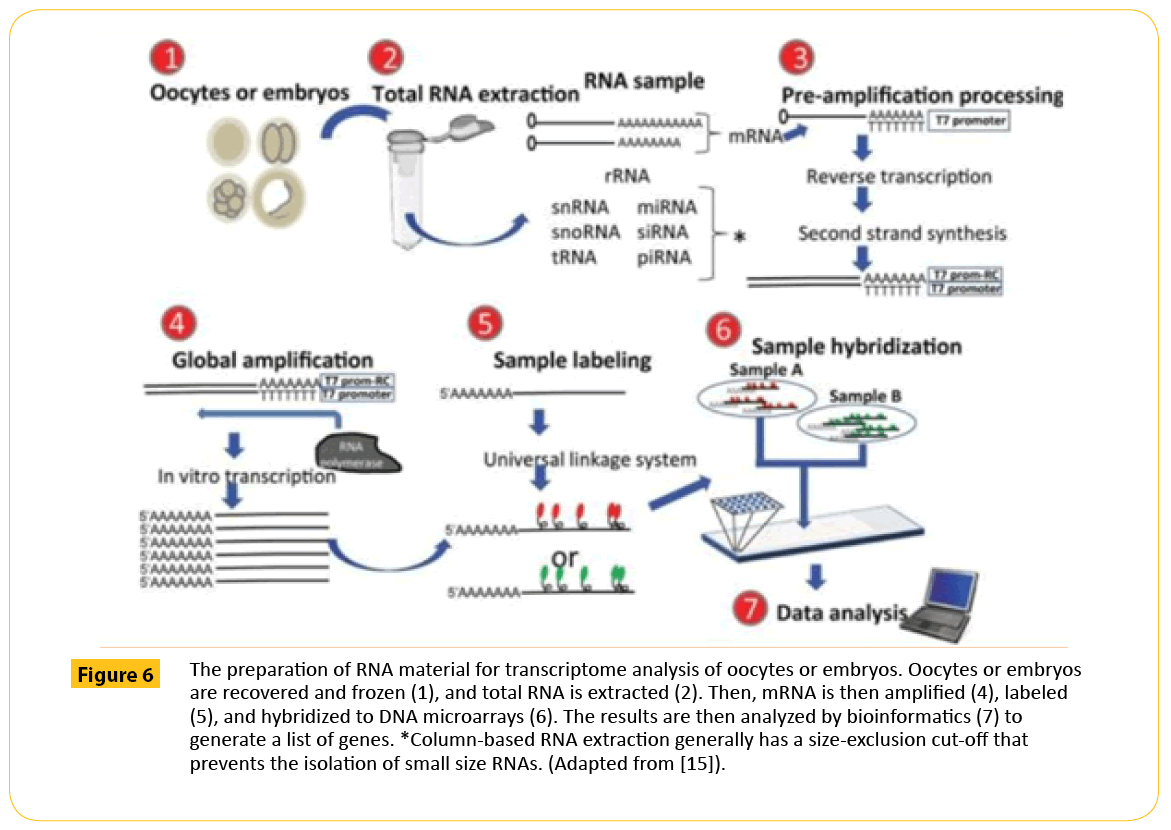
Figure 6: The preparation of RNA material for transcriptome analysis of oocytes or embryos. Oocytes or embryos are recovered and frozen (1), and total RNA is extracted (2). Then, mRNA is then ampliï¬ÂÂÂÂed (4), labeled (5), and hybridized to DNA microarrays (6). The results are then analyzed by bioinformatics (7) to generate a list of genes. *Column-based RNA extraction generally has a size-exclusion cut-off that prevents the isolation of small size RNAs. (Adapted from [15]).
Metabolomics
Metabolic process starts in an embryo from the pronuclear stage, but this is not very apparent until the embryo starts compacting. This event is followed by a switch in ATP synthesis from the carboxylic acids pyruvate and lactate as primary energy sources to glycolysis a glucose-based metabolism [73,74]. This trend continued until the blastocyst stage when glucose becomes they main metabolite of tricarboxylic acid (TCA) cycle and aerobic glycolysis [74,75].
During the process of metabolism, embryo takes up certain substances from the surrounding environment and excretes many products into the environment. The metabolic change in this environment can be measured by Ultra-micro?uorescence assay, Proton NMR and HPLC. Technologies like NMR spectroscopy, mass spectrometry (MS), which can be coupled with separation methods like gas chromatography (GC–MS), liquid chromatography (LC–MS) or HPLC–MS, and capillary electrophoresis (CE–MS). Optical spectroscopies, like Fourier transform infrared (FT- IR), near infrared (NIR) and Raman spectroscopies are excellent technologies that have improved the sensitivity of metabolic profiling [16].
If these embryos are being developed in culture media, the assessment of the spent media for substance that is taken up or released may provide information reflecting cellular activities and overall developmental potential during the culture period (Figure 7). If this information is provided in real time, the metabolic profile of the embryo may be used to select a viable one from cohort to be transferred in a fresh cycle. There is growing evidence that metabolic profiles of spent embryo culture media differ between embryos that implant and those that fail to implant and can thereby be used to predict reproductive potential (Table 6).
| Reference |
Embryo examined |
stage |
Altered associated with Improved outcome |
metabolite |
Technology used |
Outcome |
| Hardy et al. [78] |
Day 2-4 |
|
↑pyruvate uptake |
|
Ultramicrouorescence assay |
Blastocyst development |
| |
|
No association with glucose |
|
|
|
| aDay 5 |
|
↑ pyruvate uptake |
|
UltramicroÃuorescence assay |
Blastocyst development |
| |
|
↑ glucose uptake |
|
|
|
| Gott et al. [79] |
Day 2-4 |
|
↑ pyruvate uptake |
|
Ultramicroáuorescence assay |
Blastocyst development |
| |
|
↑lactate production |
|
|
|
| Day 5 |
|
↑ pyruvate uptake |
|
Ultramicrouorescence assay |
Blastocyst development |
| |
|
↑glucose uptake |
|
|
|
| |
|
↑lactate production |
|
|
|
| Conaghan et al. [80] |
Day 2-3 |
|
↓pyruvate uptake |
|
Ultramicro¡uorescence assay |
Clinical pregnancy |
| Turner et al. [81] |
Day 2 |
|
Intermediate pyruvate uptake |
|
Ultramicrouorescence assay |
Clinical pregnancy |
| Gardner et al. [82] |
Day 4 |
|
↑ pyruvate uptake |
|
Ultramicrouorescence assay |
Blastocyst development |
| |
|
↑glucose uptake |
|
|
|
| Houghton et al. [83] |
Day 2-3 |
|
↓amino acid turnover (sum of depletion and appearance) |
HPLC |
|
Blastocyst development |
| |
|
↓glutamine, arginine,methionine uptake |
HPLC |
|
|
| |
|
↓alanine and asparagine release |
|
|
|
| |
|
↓amino acid turnover (sum of depletion and appearance) |
|
|
|
| |
|
↓serine uptake |
|
|
|
| |
|
↓ alanine and glycine release |
|
|
Blastocyst development |
| Brison et al. [84] |
Day 2 |
|
↓ glycine and leucine in culture media |
HPLC |
|
Clinical pregnancy and live birth |
| |
|
↑ asparagine levels in culture media |
|
|
|
| Seli et al. [85] |
Day 3 |
|
↑glutamate levels in culture media |
Proton NMR |
|
Clinical pregnancy and live birth |
| Day 4-5 |
|
↑ Glucose level in culture media |
Microfluorimetry |
|
Clinical pregnancy |
| Pudakalakatti et al. [86] |
Day 3 |
|
↓pyruvate/alanine |
NMR spectroscopy |
|
Clinical pregnancy |
Table 6: Pyruvate, lactate, glutamate, alanine and glucose metabolism as a predictor of competency. (Adapted from [71]).
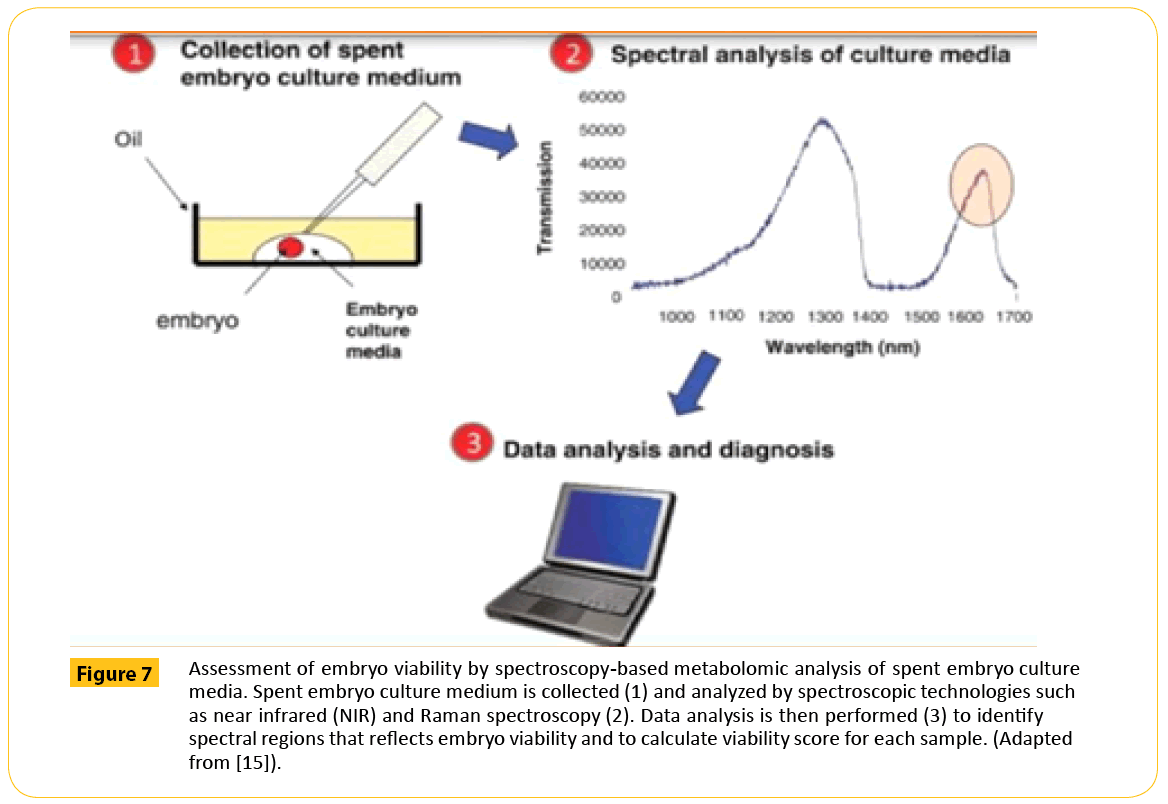
Figure 7: Assessment of embryo viability by spectroscopy-based metabolomic analysis of spent embryo culture media. Spent embryo culture medium is collected (1) and analyzed by spectroscopic technologies such as near infrared (NIR) and Raman spectroscopy (2). Data analysis is then performed (3) to identify spectral regions that reects embryo viability and to calculate viability score for each sample. (Adapted from [15]).
Despite this evidence, a lot still need to be done if metabolic profiling can be relied on for single embryo selection especially in the area of cost and expertise which could be prohibitive to most embryology laboratories. Also, some of these technologies do not produce results quickly enough to allow the information to be used clinically in the limited window of time acceptable for fresh embryo transfer. It is critical to also mention here that not all studies have shown that metabolic profiling is beneficial in selecting viable embryo [76,77]. These contrary studies have made it imperative for a well designed validation study to be undertaken in order to provide the level of evidence needed for clinical utility.
The omics of the endometrium
In every IVF cycle, the hope of the couple is to achieve conception. Even if the accuracy with which a viable embryo is selected from a cohort is 100%, it does not guarantee that conception will be achieved. This is because some failed implantation is due to the endometrium and not the embryo. Evaluating the endometrium is therefore critical to determine the endometrial receptivity before embryos are implanted. This is necessary because the endometrium provides the environment necessary for embryo to implant and for fetal growth and development [84].
The receptiveness of the endometrium is not constant throughout the cycle due to hormonal changes. This produces several events like differentiation, shedding and re- growth. Traditional methods of assessing the receptivity of the endometrium after these events have been less effective due to the complex interactions that occurs [84]. The use of omics technologies to evaluate the human endometrium may improve the understanding of biomarkers associated with receptive and non receptive endometrium.
Already, omics data has provided valuable information on genes and proteins that may be useful as biomarkers for the evaluation of endometrial receptivity [85]. The application of omic technologies to determine the proper time to transfer embryo to the endometrium is expected to improve pregnancy rate if viable embryos are transferred.
The health economics of comprehensive embryo evaluation
Single embryo transfer (SET) is the most feasible way to reduce the incidence of multiple pregnancies in IVF. Though decision to have a SET is majorly the purview of the patient, the clinician has the moral obligation to educate them on the danger of transferring more than one embryo at a time. Some patients will still opt for more than one embryo transfer even when the danger is made known. They choose this step as a way to cushion the effect of the cost of IVF and the absence of a guarantee outcome.
One way of promoting SET is vitrification. With vitrification, post thawed survival rate is very high and pregnancy is comparable to fresh embryo transfer. This means that one embryo can be transferred while others are vitrified in a cycle. The limitation of this approach is that a number of frozen embryo transfer (FET) cycles may be required to achieve a pregnancy [86]. This approach will defeat the aim of cutting cost because drugs will be required to prepare the endometrium for FET and the cost of storing the embryos too. Even when the transfer is done with a planned natural cycle, the emotional cost of getting a negative pregnancy test report is immeasurable.
There is no doubt that success rate is lower in SET and the cost of IVF is high, but what is more clear is that the cost in managing maternal and neonatal complications in multiple pregnancies could be higher. Studies have shown that higher rates of maternal and neonatal complications could lead to much higher health care costs for care of preterm births from IVF-conceived multiple gestations [87]. A comparison between SET and multiple embryo transfer in terms of cost is likely to favor SET if viable embryos are appropriately selected and transferred.
If a more comprehensive method of evaluating embryo viability like omics is introduced fully into IVF, the cohort of embryos produced in a cycle of IVF will be evaluated for viability. Only a single viable embryo will be transferred and others will be stored or discarded depending on their viability. The stored viable embryos have better guarantee than when cohort are stored based on morphological evaluation alone. This may reduce the emotional and financial cost involved in elective SET and the burden of managing multiple pregnancies.
Translating omics into a viable tool for clinical embryo selection
The use of omics in IVF is becoming more feasible due to increase in the sensitivity, resolution and throughput of the technologies that are being developed. Hopes are being raised that these technologies may contribute in the design of a non-invasive approach that will allow for single embryo transfer. To achieve this, a multi-omic approach should be adopted and designed in three stages of: Discovery, Validation and Clinical Utility.
Discovery
A multidisciplinary approach is required for the development of a multi-omic system. Best practices should be applied in sample collection, preparation and analysis during discovery if biomarkers indentified will make it beyond this phase. This is not all, the information from the sample needs to be organized properly, processed and analyzed accurately. Each of these stages is critical for the discovery of a reliable biomarker. It is therefore important that skilled professionals in the different disciplines necessary for this discovery are involved early in this search. Translational Medicine tools and structures should be properly deployed for effective integration of these disciplines. A proper analysis of a unified multi-omic data generated through this integrative approach may help identify any alteration in SNP, CNV, inversions, or transpositions, down- regulation or up-regulation of RNAs, proteins as well as metabolites that are released by an embryo. The benefit of this is that the embryo is evaluated holistically as such where a gene, protein or metabolite is poorly expressed by one omics system due to low frequency, it may be captured by another omics system where it frequency is high. This is because these alterations may occur at multiple points within the signaling pathways. Therefore integrating data among multi-omics technologies is necessary for identifying reliable biomarkers that can measure the viability of an embryo because often, one omics system only captures the activity that relates to it. For example, genomic sequencing only captures structural variations, but not RNA level. The RNA level is revealed when another omics technology such as RNA-seq is applied [88]. This may be one reason why researchers have failed to discover reproducible biomarkers that have the capability of predicting viability in embryo to approach 100% accuracy from a single omics technology till date.
A unified multi-omic data is hoped to reveal the complex interactions that occur in a biological system. To generate this unified data, there is a need to homogenize data in experiments and also the analytical protocols. This step is necessary to avoid false interpretation due to false positive or negative data set as more potential biomarkers of embryo viability are discovered. Already, large data from genomics, proteomics, transcriptomics and metabolomics have been generated from high-throughput machines. The focus now is to integrate these multi-omics data to define a point where there interactions may signify a normal or abnormal process. The first attempt in this direction was recently reported by Sun et al. [89]. They developed an integrative Pathway Enrichment Analysis Platform (iPEAP) to interrogate transcriptomics, proteomics, metabolomics and GWAS data simultaneously as a method of analyzing multipathway interactions. A platform like this may make it easier to indentify cohort of biomarker that predicts viability of an embryo from a network structure of genomics, transcriptomics, proteomics and metabolomics (Figure 8). This process may be a lot easier if known pathway information extracted from public databases such as KEGG, Reactome or PID is also utilized [90].
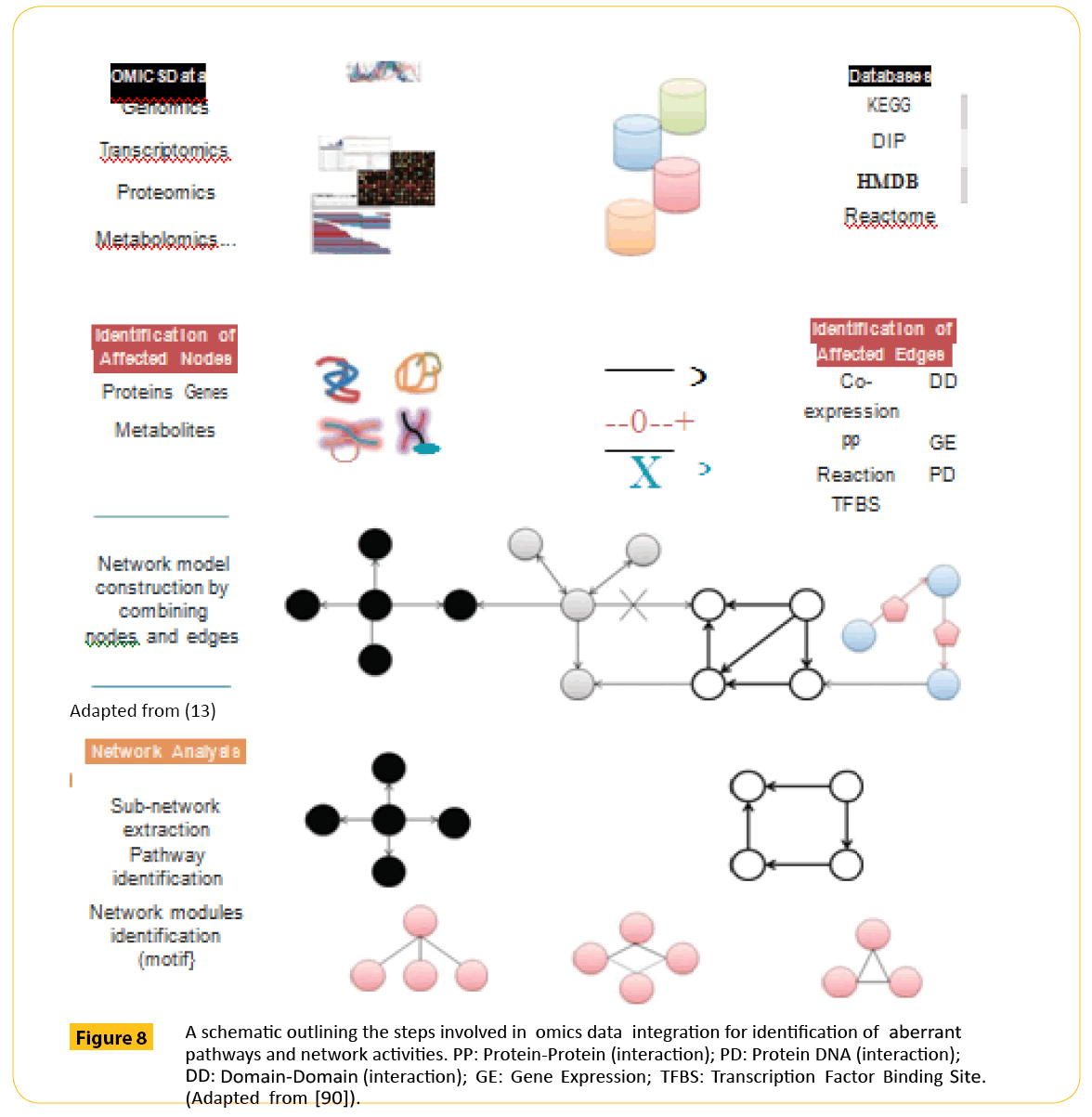
Figure 8: A schematic outlining the steps involved in omics data integration for identification of aberrant pathways and network activities. PP: Protein-Protein (interaction); PD: Protein DNA (interaction); DD: Domain-Domain (interaction); GE: Gene Expression; TFBS: Transcription Factor Binding Site. (Adapted from [90]).
It is likely that if the networks identified are decomposed into sub networks, models can be easily designed to simulate and predict network activities. Approach like this may enhance the development of valuable biomarkers that will accurately predict embryo viability.
Validation
Omics translation is driven by both data generating technologies and computational models. It is therefore important that both components be validated externally before it is implemented in the clinical selection of embryos [91]. The essence of this validation is to ensure that the omics technologies and bioinformatics tool used in measuring embryo viability are reliable. This is necessary because an embryo may be wrongly judged in the presence of biological artefacts or inappropriately applied bioinformatics method.
The difficulties in validating biomarkers from multi-omics, may not be unconnected to lack of best practices in the experiments and analytical protocols employed. This is further compounded by the fact that researchers in omics find it difficult to share data, computational models and experiences [90]. Though databases are available, practicable and easier ways to share data are necessary to improve validation of new biomarkers. Investment on suitable platforms that may enhance sharing amongst scientists and research institutions is one area that needs immediate actions to improve on validation of new biomarker. This platform will help save time and money that may be used in funding a biomarker discovery that is based on false interpretation.
False interpretation of omics data is not unexpected because the technicalities involved in the assays are complex. There are also shortcomings of the bioinformatics model and the special techniques needed to handle the samples. An error in any of these may affect the quality of the data. This makes validation important to support claims that biomarker discovered through this approach is reliable enough for clinical use in embryo selection. McShane et al. [92] recommends that the predictability of an omics biomarker should be confirmed using completely independent training data sets and the training data sets should be made to fulfill the following criteria:
1. The outcome should be blinded until after the computational procedures have been locked down and the candidate omics -based test has been applied to the samples [91].
2. Assays for the validation specimen set should be run at a different time or in a different laboratory but according to the identical assay protocol as was used for the training set [92].
3. The candidate omics -based test should be defined precisely, including the molecular measurements, the computer code, the computational procedures, and the intended clinical use of the test, in anticipation of the test validation phase [92].
These criteria if properly implemented have the potential of making validation reproducible when done in any other laboratory.
Clinical utility
Omics technologies have helped to generate a number of high profile biomarkers that can be useful in selecting embryos that are viable from those that are not. The problem is that most of them are launched into clinical trial without thorough consideration of factors that could influenced their outcome, as well as the ethical, legal, and regulatory issues involved. Biomarkers discovered through multi-omics systems are subjected to a different regulatory framework from drugs [91]. In the US for example, there are two ways in which biomarkers discovered through multi-omic system can be approved for use clinically to screen embryos.
1. Through FDA as commercial test kits
2. Through certify CLIA laboratory as laboratory-developed tests (LDTs).
Most biomarker test kits discovered through omics technologies are developed as LDT to bypass the hurdles of FDA. The disadvantage of this route is that the test can only be use to correlate the result with the outcome but not to specify the diagnosis [93]. This is because the discovery and validation phase is not reviewed by regulatory authority as such it safety and effectiveness cannot be confirmed. Though FDA is statutory empowered to oversee all laboratory tests that is used in clinical decision making, it has paid attention only to those considered to be of high complexity and of high risk [91]. A more defined regulatory approach is necessary for biomarkers developed through multi-omic system to address the lack of transparency in the LDTs process.
For an omic biomarker to qualify for clinical utility, it must have been properly validated. Many promising omics failed to go beyond the validation phase despite the huge investment made. Some reasons why many of them failed are related to study design, patient selection, sample integrity, data analysis and management. To address these issues, McShane et al. [92] developed a criteria checklist (Table 7). These criteria if fully implemented could help develop a more efficient, reliable and transparent process to move omics biomarkers from mere promising research results to clinically useful tests that will permit the transfer of single viable embryo.
| DomainCriteria |
| Specimen issues |
1. Establish methods for specimen collection and processing and appropriate storage conditions to ensure the suitability of specimens for use with the omics test. |
| 2. Establish criteria for screening out inadequate or poor-quality specimens or analytes isolated from those specimens before performing assays. |
| 3. Specify the minimum amount of specimen required. |
| 4. Determine the feasibility of obtaining specimens that will yield the quantity and quality of isolated cells or analytes needed for successful assay performance in clinical settings. |
| Assay issues |
5. Review all available information about the standard operating procedures (SOPs) used by the laboratories that performed the omics assays in the developmental studies, including information on technical protocol, reagents, analytical platform, assay scoring, and reporting method, to evaluate the comparability of the current assay to earlier versions and to establish the point at which all aspects of the omics test were definitively locked down for final validation. |
| 6. Establish a detailed SOP to conduct the assay, including technical protocol, instrumentation, reagents, scoring and reporting methods, calibrators and analytical standards, and controls. |
| 7. Establish acceptability criteria for the quality of assay batches and for results from individual specimens. |
| 8. Validate assay performance by using established analytical metrics such as accuracy, precision, coefficient of variation, sensitivity, specificity, linear range, limit of detection, and limit of quantification, as applicable. |
| 9. Establish acceptable reproducibility among technicians and participating laboratories and develop a quality assurance plan to ensure adherence to a detailed SOP and maintain reproducibility of test results during the clinical trial. |
| 10. Establish a turnaround time for test results that is within acceptable limits for use in real-time clinical settings. |
| Model development, |
11. Evaluate data used in developing and validating the predictor model to check for accuracy, completeness, and outliers. |
| specification, and preliminary performance evaluation |
Perform retrospective verification of the data quality if necessary. |
| 12. Assess the developmental data sets for technical artifacts (for example, effects of assay batch, specimen handling, assay instrument or platform, reagent, or operator), focusing particular attention on whether any artifacts could potentially influence the observed association between the omics profiles and clinical outcomes. |
| 13. Evaluate the appropriateness of the statistical methods used to build the predictor model and to assess its performance. |
| 14. Establish that the predictor algorithm, including all data pre-processing steps, cutpoints applied to continuous variables (if any), and methods for assigning confidence measures for predictions, are completely locked down (that is, fully specified) and identical to prior versions for which performance claims were made. |
| 15. Document sources of variation that affect the reproducibility of the final predictions, and provide an estimate of the overall variability along with verification that the prediction algorithm can be applied to one case at a time. |
| 16. Summarize the expected distribution of predictions in the patient population to which the predictor will be applied, including the distribution of any confidence metrics associated with the predictions. |
| 17. Review any studies reporting evaluations of the predictor’s performance to determine their relevance for the setting in which the predictor is being proposed for clinical use. |
| 18. Evaluate whether clinical validations of the predictor were analytically and statistically rigorous and unequivocally blinded. |
| 19. Search public sources, including literature and citation databases, journal correspondence, and retraction notices, to determine whether any questions have been raised about the data or methods used to develop the predictor or assess its performance, and ensure that all questions have been adequately addressed. |
| Clinical trial design |
20. Provide a clear statement of the target patient population and intended clinical use of the predictor and ensure that the expected clinical benefit is sufficiently large to support its clinical utility. |
| 21. Determine whether the clinical utility of the omics test can be evaluated by using stored specimens from a completed clinical trial (that is, a prospective–retrospective study). |
| 22. If a new prospective clinical trial will be required, evaluate which aspects of the proposed predictor have undergone sufficiently rigorous validation to allow treatment decisions to be influenced by predictor results; where treatment assignments are randomized, provide justification for equipoise. |
| 23. Develop a clinical trial protocol that contains clearly stated objectives and methods and an analysis plan that includes justification of sample size; lock down and fully document all aspects of the omics test and establish analytical validationof the predictor. |
| 24. Establish a secure clinical database so that links among clinical data, omics data, and predictor results remain appropriately blinded, under the control of the study statistician. |
| 25. Include in the protocol the names of the primary individuals who are responsible for each aspect of the study. |
| Ethical, legal, andregulatory issues issues that are relevant to the conduct of the trial. |
26. Establish communication with the individuals, offices, and agencies that will oversee the ethical, legal, and regulatory |
| 27. Ensure that the informed consent documents to be signed by study participants accurately describe the risks and potential benefits associated with use of the omics test and include provisions for banking of specimens, particularly to allow for ‘bridging studies’ to validate new or improved assays. |
| 28. Address any intellectual property issues regarding the use of the specimens, biomarkers, assays, and computer software used for calculation of the predictor. |
| 29. Ensure that the omics test is performed in a Clinical Laboratory Improvement Amendments-certified laboratory if the results will be used to determine treatment or will be reported to the patient or the patient’s physician at any time, evenafter the trial has ended or the patient is no longer participating in the study. |
| 30. Ensure that appropriate regulatory approvals have been obtained for investigational use of the omics test. If a prospective trial is planned in which the test will guide treatment, consider a pre-submission consultation with the US Food and Drug Administration. |
Table 7: Criteria for the use of omics-based predictors in National Cancer Institute-supported clinical trials. (Adapted from [92]).
Future direction
The mainstay of morphology method for assessing human embryo viability is the simplicity and low cost. The current approach in omics biomarker discovery is broad based hence the high cost and technical complexities. Already, some of these biomarkers have shown correlation with the state of health of human embryo. It is likely that a cohort of biomarkers that can tell when an embryo is viable or not from gene, protein, metabolites and RNA detectable from the spent media they are cultured in will soon be discovered. Once these biomarkers are discovered, efforts should be made to translate it into a multiomic platform that will allow for the detection of multi-omic parameter from a single spent media sample. The creation of such a platform may encourage IVF clinics to abandon the current morphology method and adopt it use because of simplicity, rapid turnaround time and cost effectiveness. This diagnostic platform may be used in the IVF laboratory, to select the most appropriate embryo.
Elementary description of the platform
The platform should be like a portable dry clinical chemistry analyzer. It should be designed in the form of a desk top system. The system should consist of the analyzer and the bioinformatics component connected by a cord at the back. Another cord should lead to a bigger monitor where the results will be displayed. A printer may be attached to the system if need be.
2. The analyzer component should be able to detect and quantify all parameters that correlate with viability from genomics, proteomics, metabolomics and transcriptomics from test strips through the principle of reflectance photometry. The analyzer should have ports where the multi-omic test strips will be inserted simultaneously
3.A bioinformatics component should be developed to analyze results from the multi- omic parameters from a spent media at the same time and provide information about global health status of the embryo from which the spent media was taken. The information about the embryo should be rapidly displayed on a monitor.
4. The test strip should be developed with antibodies recovered after immunizing a host model with a powerful adjuvant. The antibodies recovered should be immobilized onto a support, and should be able to selectively capture biomarkers antigen that correlates with viability directly from spent media. Information about the omic biomarkers from the spent media should be readily captured by the analyzer from the strip once inserted and transferred to the bioinformatics model for analysis.
Antibody based omics is a low cost way of bringing omics into the embryology laboratories. Antibody are specific in action, they can be developed to interrogate the genome, proteome, transcriptome and metabolome. Producing antibodies against metabolites may pose a considerable challenge but not impossible as antibodies against cocaine metabolites already exist. A strategy like this may allow for laboratory testing just before embryos are transferred to inform decision making. When such a platform is eventually developed, it should be properly validated through a transparent clinical trial before it application in clinical selection of embryos
Conclusion
Translational Medicine is being introduced as the discipline that will fill in the existing gap between laboratory-based research and patient treatment. It is believed that this gap can be bridged if knowledge gained from basic science research is applied into clinical practice and outcome of this research in the clinic is injected back into basic science research. This line of thought is a welcome development in IVF considering the lower success rate seen in single embryo transfers compared to multiple embryo transfers. Multiple embryo transfer is being discouraged because of the risk posed by multiple pregnancies to the mother and health of the babies. A lot of investment has been made on technologies that would select a single viable embryo to encourage SET. This investment ranges from technologies used for morphology based test like embryoscope to genetic testing technologies including FISH, qPCR, aCGH, array SNP and NGS. Other technologies that could assess embryo metabolism, protein and RNA profile are available. Despite this remarkable milestone, result from SET has not improved significantly and fertility challenged couples are still being exposed to the risk of multiple pregnancies. Considerable investment and time have been inputted into the search for a suitable technology capable of selecting a single viable embryo, the next logical step to move the research that has been done on the bench towards defining a viable embryo that will improve on the outcome of SET is translational medicine (TM). Infusing TM in IVF is now necessary because large amount of data about the sequence of DNA, global gene expression, proteomics and metabolomics of an embryo is already known. These discoveries need to be translated into the IVF clinics in order to improve on the selection of viable embryos from a cohort. The ability of TM to move this translation is hinged on the fact that TM presents an interdisciplinary approach, mobilizes scientific and regulatory disciplines as well as technologies like omics to discover biomarkers, computational biology and bioinformatics for integration of this data, biostatistics for multiple testing and regulatory knowledge for timely introduction of this technology to the clinics.
4887
References
- Wang J, Sauer MV (2006) In vitro fertilization (IVF): a review of 3 decades of clinical innovation and technological advancement. TherClin Risk Manag 2: 355-364.
- Papanikolaou EG, D'haeseleer E, Verheyen G, Van de Velde H, Camus M, et al. (2005) Live birth rate is significantly higher after blastocyst transfer than after cleavage-stage embryo transfer when at least four embryos are available on day 3 of embryo culture, A randomized prospective study. Hum Reprod 20: 3198-3203.
- FauserBC,DiedrichK, BouchardP,DomínguezF,etal. (2011) EvianAnnualReproduction(EVAR) WorkshopGroup 2010,Contemporarygenetictechnologiesand female reproduction.HumReprodUpdate17:829-847.
- https://www.asrm.org/uploadedFiles/Affiliates/SART/Members/Forms/Embryo%20Morphology.pdf
- Scott L, Berntsen J, Davies D, Gundersen J, Hill J, et al. (2008) Symposium: innovative techniques in human embryo viability assessment. Human oocyte respiration-rate measurement-potential to improve oocyte and embryo selection? Reprod Biomed Online 17: 461-469.
- Palini S, Galluzzi L, De Stefani S, Bianchi M, Wells D, et al.(2013) Genomic DNA in human blastocoele fluid. Reprod Biomed Online 26: 603-610.
- Cohen J, Grudzinskas G, Johnson MH (2013) Embryonic DNA sampling without biopsy: the beginnings of non-invasive PGD? 26: 520-521.
- Fragouli E, Lalioti MD, Wells D (2014) Thetranscriptome of follicular cells: biological insights and clinical implications for the treatment of infertility.Hum Reprod Update 20: 1-11.
- Braude P (2013) Symposium: Selecting the “best” embryos: prospects for improvement. Reproductive BioMedicine Online 27: 644-653.
- Northrop LE, Treff NR, Levy B, Scott Jr RT (2010) SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocysts. Mol Hum Reprod 16: 590-600.
- SeliE,RobertC, SirardMA (2010)OMICSinassistedreproduction:possibilitiesand pitfalls.Molecular HumanReproduction16:513-530.
- Dominguez F,Pellicer A,Simon C (2009)TheHumanEmbryoProteome. Reproductive Science16:188-190
- BrownTA (2002)Genomes.(2ndedn)Oxford:Wiley-Liss;Chapter3, TranscriptomesandProteomes.
- BrisonDR,HoughtonFD,FalconerD,RobertsSA,HawkheadJ,et al. (2004) Identificationofviableembryos in IVF bynon-invasivemeasurementofaminoacid turnover. HumReprod19: 2319-2324.
- SeliE,BotrosL, SakkasD,BurnsDH (2008) Non-invassivemetabolomics profilingof culturemedia using protonNMRcorrelateswithreproductivepotential ofembryosin womenundergoinginvitrofertilization.FertilSteril90: 2183-2189
- Gardner DK, Wale PL, Collins R, Lane M (2011) Glucose consumption of single post- compaction human embryos is predictive of embryo sex and live birth outcome. Hum Reprod 26: 1981-1986.
- Pudakalakatti SM, Uppangala S, D'Souza F, Kalthur G, Kumar P, et al. (2013) NMR studies of preimplantation embryo metabolism in human assisted reproductive techniques: a new biomarker for assessment of embryo implantation potential. NMR Biomed 26: 20-27.
- AlphaScientistsinReproductiveMedicineandESHRESpecialInterestGroup of Embryology(2011) TheIstanbulconsensusworkshoponembryoassessment:proceedingsof an expertmeeting.Reprod Biomed Online22: 632-646.
- Kirkegaard K,AgerholmIE,Ingerslev HJ (2012) Time-lapsemonitoring asatool for clinical embryoassessment.HumReprod27: 1277-1285.
- BoltonVN,Hawes SM,TaylorCT,ParsonsJH (1989)Developmentofspare humanpre- implantationembryosin-vitro:ananalysisofthecorrelations amonggrossmorphology, cleavage rates, anddevelopmentto theblastocyst. J In VitroFert Embryo Transf6:30-35.
- GardnerDK,VellaP,LaneM, WagleyL,SchlenkerT, et al. (1998) Cultureand transferofhumanblastocystsincreasesimplantationratesandreducestheneed for multiple embryo transfers.FertilSteril69:84-88
- WongC,LoewkeK,BossertN,BehrB,DeJongeC,etal. (2010)Non-invasive imagingofhumanembryosbeforeembryonicgenomeactivationpredictsdevelopment to the blastocyststage.NatBiotechnol28: 1115-1121
- Kirkegaard K, Kesmodel US, Hindkjær JJ, Ingerslev HJ (2013) Time-lapse parameters as predictors of blastocyst development and pregnancy outcome in embryos from good prognosis patients: a prospective cohort study. Hum Reprod 28: 2643-2651.
- HerreroJ,MeseguerM (2013)Selectionofhighpotentialembryosusingtime-lapse imaging:theera ofmorphokinetics.FertilandSteril99: 0015-0282.
- Chen AA, Tan L, Suraj V, ReijoPera R, Shen S (2013) Biomarkers identi?ed with time- lapse imaging: discovery, validation, and practical application. FertilSteril 9: 1035-1043.
- Swain JE (2013) Could time-lapse embryo imaging reduce the need for biopsy and PGS? J Assist Reprod Genet 3:1081-1090.
- SimpsonJL (2012) Preimplantationgenetic diagnosistoimprove pregnancy outcomesin subfertility.BestPractResClinObstetGynaecol26: 805-815.
- Munné S (2012) Preimplantation Genetic Diagnosis for Aneuploidy and Translocations Using Array Comparative Genomic Hybridization. Curr Genomics 13: 463-470.
- Santiago Munné (2012) Preimplantation Genetic Diagnosis for Aneuploidy and TranslocationsUsingArrayComparativeGenomicHybridizationCurrGenomics 13: 463-470.
- Zamora S, Clavero A, Gonzalvo MC, de Dios Luna Del Castillo J, Roldán-Nofuentes JA, et al. (2011) PGS-FISH in reproductive medicine and perspective directions for improvement: a systematic review. J Assist Reprod Genet 28: 747-757.
- BlockeelC,SchutyserV,VosA (2008)Prospectivelyrandomizedcontrolledtrialof PGS in IVF/ICSI patients with poor implantation.Reprod Biomed Online17: 848-854.
- DebrockS,MelotteC,SpiessensC (2010)Preimplantationgeneticscreeningfor aneuploidyofembryosafterinvitrofertilizationinwomenagedatleast35years:a prospectiverandomized trial.FertilSteril93: 364-373.
- HardarsonT, Hanson C, Lundin K (2008) Preimplantationgenetic screening in womenofadvanced maternalagecausedadecreaseinclinicalpregnancy rate:a randomizedcontrolledtrial.HumReprod23: 2806-2812.
- JansenRP,BowmanMC,BoerKA,LeighDA,LiebermanDB, et al. (2008) What nextforpreimplantationgeneticscreening(PGS)?Experiencewithblastocystbiopsy andtestingfor aneuploidy.HumReprod23: 1476-1478.
- MastenbroekS, TwiskMV,Echten-Arends JV,Sikkema-Raddatz B,Johanna C,et al. (2007) In vitro fertilization with preimplantationgenetic screening.N Engl J Med357: 9-17.
- MersereauJE,PergamentE,Zhang X,MiladMP (2008) Preimplantationgenetic screening toimproveinvitrofertilizationpregnancyrates:aprospectiverandomizedcontrolled trial.FertilSteril90: 1287-1289.
- Meyer LR, KlipsteinS,HazlettWD,NastaT,Mangan P, et al. (2009) A prospective randomizedcontrolled trialofpreimplantationgeneticscreeninginthe“goodprognosis” patient.FertilSteril91: 1731-1738.
- SchoolcraftWB,Katz-JaffeMG,StevensJ,RawlinsM,MunneS (2009) Preimplantationaneuploidy testingforinfertilepatientsof advancedmaternalage:arandomized prospective trial.FertilSteril92: 157-162.
- StaessenC,PlatteauP,AsscheE (2004)Comparisonofblastocysttransferwithor without preimplantationgenetic diagnosis for aneuploidy screening in couples with advanced maternal age: a prospective randomized controlled trial.Hum Reprod19: 2849-2858.
- StaessenC,Verpoest W,DonosoP (2008)Preimplantationgeneticscreeningdoes not improve delivery rate in women under the age of 36 following single-embryo transfer.HumReprod23: 2818-2825.
- StevensJ, WaleP,SurreyES,Schoolcraft WB (2004)Isaneuploidyscreeningforpatients aged 35 or over beneficial? A prospective randomized trial.FertilSteril82: 249.
- ScottR, TaoX,TaylorD,Ferry KM,TreffNR (2010) A prospectiverandomizedcontrolled trialdemonstratingsignificantly increasedclinicalpregnancyrates following24 chromosomeaneuploidyscreening:biopsyandanalysisonday5withfreshtransfer. FertilSteril94: S2
- Gutierrez-MateoC,CollsP,Sanchez-GarciaJ,EscuderoT,PratesR, et al. (2011) Validation of microarray comparative genomic hybridization for comprehensivechromosomeanalysis ofembryos.FertilSteril95: 953-958.
- FioretinoF,SpizzichinoL,Bono S,BirricikA,KokkaliG, et al. (2011) PGD for reciprocal and Robertsonian translocation using array comparative genomic hybridization. Hum Reprod26: 1925-1935.
- Hodes-Wertz B, Grifo J, Ghadir S, Kaplan B, Laskin CA, et al. (2012) Idiopathic recurrent miscarriage is caused by aneuploid embryos. FertilSteril 98: 675-680.
- Yang Z, Liu J, Collins G, Salem S, Liu X, et al. (2012) Selection of single blastocysts for fresh transfer via stand morphology assessment alone and with array CGH for good prognosis IVF patients: results from a randomized pilot study. MolCytogenet 5: 24.
- Handyside AH (2011) PGD and aneuploidy screening for 24 chromosome by genome-wide SNP analysis: seeing the woods and the trees. Reprod Biomed Online 23: 686-691.
- Yang Z, Salem SA, Liu X, Kuang Y, Salem RD, et al. (2013) Selection of euploid blastocysts for cryopreservation with array comparative genomic hybridization (aCGH) results in increased implantation rates in subsequent frozen and thawed embryo transfer cycles. MolCytogenet 6: 32.
- Treff NR, Scott RT Jr (2012) Methods for comprehensive chromosome screening of oocytes and embryos: capabilities, limitations, and evidence of validity. J Assist Reprod Genet 29: 381-390.
- LeCaignecC,SpitsC,SermonK,DeRyckeM,ThienpontB, et al. (2006)Single-cellchromosomalimbalances detectionbyarrayCGH.Nucleic AcidsRes34:e68
- Wells D, Alfarawati S, Fragouli E (2008) Use of comprehensive chromosomal screening for embryo assessment: microarrays and CGH. Mol Hum Reprod 14: 703-710
- MartinJ,CerveroA,MirP,Martinez-ConejeroJA,PellicerA, et al. (2013)SimonC.Theimpact ofnext-generationsequencing technology onpreimplantationgenetic diagnosis and screening.FertilSteril99:1054-1061.
- O?Leary T, Heindryckx B, Lierman S, Van der Jeught M, Duggal G, et al. (2013) Derivation of human embryonic stem cells using a post- inner cell mass intermediate.Nat Protoc 8: 254- 264.
- Brooke Hodes-Wertz, Jamie Grifo, ShahinGhadir, Brian Kaplan, Carl ALaskin, et al. (2012) Idiopathic recurrent miscarriage is caused mostly by aneuploid embryos. FertilSteril 98: 675-680.
- Schoolcraft WB, Surrey E, Minjarez D, Gustofson RL, Scott Jr, et al. (2012) Comprehensive chromosome screening (CCS) with vitrification results in improved clinical outcome in women> 35 years: a randomized control trial. Fertility and Sterility 98: S1.
- Forman EJ, Hong KH, Ferry KM, Tao X, Taylor D, et al. (2013) In vitro fertilization with single euploid blastocyst transfer: a randomized controlled trial. FertilSteril 100: 100-7.e1.
- YangZ, LiuJ,CollinsGS,SalemSA,LiuX, et al. (2012)Selectionof singleblastocystsforfreshtransferviastandardmorphologyassessmentaloneand with array CGH for good prognosis IVF patients: results from a randomized pilot study.Molecular cytogenetics5: 1-8.
- Rubio C, Rodrigo L, Mir P, Mateu E, Peinado V, et al. (2013) Use of array comparative genomic hybridization (array- CGH) for embryo assessment: clinical results. FertilSteril 99: 1044-1048.
- BettegowdaA,LeeK, SmithGW (2007)Cytoplasmicandnucleardeterminantsofthe maternal-to- embryonictransition.ReprodFertilDev20: 45-53.
- Leese HJ, Sturmey RG, Baumann CG, McEvoy TG (2007) Embryo viability and metabolism: obeying the quiet rules. Hum Reprod 22: 3047-3050.
- Braude P,BoltonV, MooreS (1988)Human gene expression first occurs betweenthefour- andeight-cellstages of pre-implantationdevelopment.Nature 332:459-461.
- Katz-Jaffe MG, McReynolds S, Gardner DK, Schoolcraft WB (2009) The role of proteomics in defining the human embryonic secretome. Mol Hum Reprod 15: 271-277.
- DominguezF,GadeaB,MercaderA (2010) Embryologicoutcomeandsecretomeprofileofimplantedblastocystsobtainedafter coculturein humanendometrial epithelial cells versus thesequential system.FertilSteril93: 774-782
- Micheel, Nass, OmennSJ (2012) Evolution of translational omics: lessons learned and the path forward. National Academies Press, Washington DC.
- Dobson AT, Raja R, Abeyta MJ, Taylor T, Shen S, et al. (2004) The unique transcriptome through day 3 of human pre-implantation development. Hum Mol Genet 13: 1461-1470.
- O?Leary T, Heindryckx B, Lierman S, Van der Jeught M, Duggal G, et al. (2013)Derivation of human embryonic stem cells using a post- inner cell mass intermediate. Nat Protoc 8: 254-264.
- Morozova O, Hirst M, Marra MA (2009) Applications of new sequencing technologies for transcriptome analysis Annu Rev Genomics. Hum Genet 10: 135-151.
- BrinsterRL (1965)Studiesonthedevelopmentofthemouseembryosinvitro.II.The effectof energysource. J. Exp.Zool158:59-68.
- Gardner DK (1998) Changes inrequirementsandutilizationof nutrientsduringmammalian preimplantationembryo development and their significance in embryo culture. Theriogenology49: 83-102
- GardnerDK,LeeseHJ (1986)Non-invasivemeasurementofnutrientuptakeby single culturedpre-implantationmouseembryos.HumReprod1: 25
- Vergouw CG, Kieslinger DC, Kostelijk EH, Botros LL, Schats R, et al. (2012) Day 3 embryo selection by metabolomic profiling of culture medium with near-infrared spectroscopy as an adjunct to morphology: a randomized controlled trial. Hum Reprod 27: 2304-2311.
- Vergouw CG, Heymans MW, Hardarson T, Sfontouris IA, Economou KA, et al. (2014) No evidence that embryo selection by near-infrared spectroscopy in addition to morphology is able to improve live birth rates: results from an individual patient data meta-analysis. Hum Reprod 29: 455-461.
- HardyK,HooperMA,HandysideAH,RutherfordAJ, WinstonRM, et al. (1989) Noninvasivemeasurementofglucoseandpyruvateuptakeby individualhumanoocytes andpreimplantationembryos.Hum Reprod4: 188-191
- GottAL,Hardy K,WinstonRM,LeeseHJ (1990)Non-invasivemeasurementofpyruvate andglucoseuptakeandlactateproductionby singlehumanpreimplantationembryos. HumReprod5: 104-108
- ConaghanJ,HardyK,Handyside AH,WinstonRM, LeeseHJ, et al. (1993) Selectioncriteriafor human embryo transfer: acomparisonof pyruvate uptakeand morphology.J Assist ReprodGenet 10: 21-30
- Turner K,MartinKL,Woodward BJ,LentonEA,LeeseHJ, et al. (1994) Comparison ofpyruvate uptake byembryos derivedfromconception andnon-conceptionnatural cycles.Hum Reprod9: 2362-2366.
- Gardner DK, Lane M, Stevens J, Schoolcraft WB (2001) Noninvasive assessment of human embryo nutrient consumption as a measure of developmental potential. FertilSteril 76: 1175-1180.
- Houghton FD,HawkheadJA,HumphersonPG,Hogg JE,BalenAH, et al. (2002) Non-invassiveaminoacid turnover predictshumanembryodevelopmental capacity.Hum Reprod17: 999-1005.
- Charalampos S, ParaskeviV,AngelikiV, TerezaV,IliodromitiZ,et al. (2012) Endometrium inImprovingIVFOutcomes:AssessingitsBehaviorandMakinganAlly fromanEnemy. ReprodSys Sexual Disorders1: e105.
- Haouzi D, DechaudH, AssouS, De VosJ, HamamahS (2012)Insights into human endometrialreceptivity fromtranscriptomicandproteomicdata. Reproductive BioMedicineOnline 24:23-34.
- De Sutter P, Gerris J, Dhont M (2002) A health-economic decision-analytic model comparing double with single embryo transfer in IVF/ICSI. Hum Reprod 17: 2891-2896.
- Fiddelers AA, Severens JL, Dirksen CD, Dumoulin JC, Land JA, et al. (2007) Economic evaluations of single- versus double-embryo transfer in IVF. Hum Reprod Update 13: 5-13.
- Emily A. Vucic, Kelsie L.Thu, Keith Robison, Leszek A. Rybaczyk, Raj Chari, et al. (2012) Translating cancer “omics” to improved outcomes.Genome Res 22: 188-195.
- Sun H, Wang H, Zhu R, Tang K, Gong Q, et al. (2014) iPEAP: integrating multiple omics and genetic data for pathway enrichment analysis. Bioinformatics 30: 737-739.
- Wang J, Zhang Y, Marian C, Ressom HW (2012) Identification of aberrant pathways and network activities from high-throughput data. Brief Bioinform 13: 406-419.
- IOM (Institute of Medicine) 2012 Evolution of Translational Omics: Lessons Learnedandthe PathForward. Washington,DC:TheNational AcademiesPress.
- McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, et al. (2013) Criteria for the use of omics-based predictors in clinical trials: explanation and elaboration. BMC Med 11: 220.
- https://www.marsdd.com/dms/entrepreneurtoolkit/RegulatoryPDFs/Pathway_to_Commercialization_for_an_In_Vitro_Diagnostic_in_the_US.pdf

