Keywords
Alzheimer’s disease; Angiotensin IV; Nle1-Angiotensin IV; Dihexa; AT4 receptor subtype; Hepatocyte growth factor; Met receptor
Introduction
Neurodegenerative diseases are characterized by progressive neuron losses in specific brain regions that impact cognitive, sensory and/or motor functioning. These disorders include Alzheimer’s disease (AD), Parkinson’s disease, amyotrophic lateral sclerosis and Huntington’s disease. The destructive nature of the damage inflicted by these diseases results in a loss of patient dignity and self-worth and negatively impacts family members and friends. The current number of AD patients in the U.S. is 4.5-5.0 million [1,2] with an estimated 16 million worldwide [3]. Present annual treatment and care costs in the U.S. are $70-100 billion [4,5] and in excess of $600 billion worldwide [6]. De la Torre [7] has estimated that a treatment that delays the onset of symptoms by 5 years would reduce the number of AD patients by upwards of 50%. Thus, significant efforts are presently directed at designing drugs capable of slowing the progression of disease symptoms, with the ultimate goal of halting and reversing the underlying causes of AD.
The long-standing theory regarding the pathogenesis of AD is the “amyloid cascade hypothesis” [8,9]. This theory assumes that AD is due to the cellular deposition of insoluble amyloid β (Aβ) protein fragments arising from amyloid precursor protein proteolysis. It is suggested that dysfunction between Aβ production and clearance causes damaging accumulations of cellular Aβ, coupled with hyperphosphorylation of neuronal tau protein resulting in neurofibrillary tangle formation [10,11]). Other researchers have proposed that these cellular changes are the result of “chronic cerebral hypoperfusion” facilitated by hypertension, atherosclerosis, diabetes, hypercholesterolemia and aging [11-14]. These clinical disorders can lead to hypoperfusion which in turn results in insufficient oxygen and glucose delivered to the brain [15]. Such a condition can damage parenchymal cells and disrupt blood-brain barrier (BBB) glucose transport [16,17] leading to oxidative stress [13,18,19], inflammation [20-22] and alterations in nitric oxide levels [23-25]. It is proposed that such a disruption of BBB permeability produces a “vicious circle” in which incremental reductions in cerebral perfusion accelerates the neurodegenerative process thus facilitating further drops in cerebral perfusion [11,26,27]. De la Torre [28,29] has long argued that the accumulating consequences of cerebral hypoperfusion precedes the appearance of clinical symptoms by many years. The goal of providing an effective treatment for AD has been elusive due to the complexity of the disease process and resulting inability to identify reliable biomarkers prompting competing hypotheses regarding etiology. In addition, some AD diagnostic indicators are present in other clinical conditions including vascular disease, frontotemporal dementia, Parkinson’s disease and HIV infection induced dementia, as well as normal aging [30-33]. These factors make drug development a very challenging task. However, a treatment designed to delay the onset of AD symptoms would maintain the patient’s quality of life, significantly reduce health care costs, and may be possible in the near future.
A step in this direction would be a treatment strategy designed to offset neuron losses by stimulating synaptogenesis in existing neurons and the formation of new functional neurons thus slowing disease progression. The neurotrophic agents capable of facilitating synaptogenesis and neurogenesis include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4 [34,35]. To date BDNF has received the most attention [36]. Our laboratory has focused on an overlooked neurotrophic factor, hepatocyte growth factor (HGF), and found it to be more potent than BDNF when activated by angiotensin IV (AngIV) and AngIVbased analogs [37]. These analogs allosterically mimic dimerization/ activation, a prerequisite to binding at the Type 1 tyrosine kinase receptor Met [38,39]. This review initially describes current FDA approved drugs to treat AD, the renin-angiotensin system’s (RAS) role in memory formation, followed by a description of the HGF/ Met system. We conclude with details concerning the development and testing of AngIV-based analogs that activate the brain HGF/Met receptor system and show promise as anti-dementia agents in animal models of AD. We also detail the limitations of these molecules.
Available Treatments for Alzheimer’s Disease
At present there are no adequate drug treatments available for AD. Current FDA approved drugs fall under two classes: 1) Cholinesterase inhibitors such as Razsadyne, Exelon, Cognex and Aricept that maintain a 75% market share [40,41]. These drugs are designed to disrupt the degradation of acetylcholine and thus extend the half-life and availability of this neurotransmitter acting at central cholinergic muscarinic and nicotinic receptors. Although the original research conducted on animal models was promising concerning the role of acetylcholine in memory consolidation and retrieval, the ability of such drugs to delay symptom in AD patients is modest at best [42]. 2) Namenda (memantine HCl) received FDA approval in 2004 and acts as an N-methyl-D-aspartate (NMDA) receptor antagonist intended to limit glutamate excitotoxicity and resulting neuronal damage [40,43,44]. Namenda does appear to slow the progression of disease symptoms in some patients, particularly when given in combination with acetylcholinesterase inhibitors [45,46], but does not impact the underlying neuropathology of this disease.
In light of the “amyloid cascade hypothesis” considerable research effort has focused on the accumulation of insoluble brain Aβ protein as a basic cause of AD [47,48]. Thus, a major goal of drug development has been to attenuate and possibly block the production and deposition of Aβ1-42 [49,50]. Unfortunately Phase III clinical trials of these drugs were prematurely terminated when some mild-to-moderate AD patients evidenced reduced cognitive functioning as compared with patients receiving placebo [51,52]. This treatment approach falls short and modified and/or new strategies must be developed [53,54]. Another current approach is concerned with tau accumulations at synapses and the role of tau phosphorylation and acetylation [55,56]. If the chronic “cerebral hypoperfusion hypothesis” is correct then brain neuronal accumulations of Aβ1-42 and tau proteins must be considered downstream consequences rather than causes of AD [48]. Further, a potential treatment strategy must include mechanisms to facilitate and maintain cerebral blood flow and encourage neuron replacement in damaged brain structures. As outlined in the next section the angiotensin system may offer such remedies.
The Angiotensin IV/AT4 Receptor System and Memory
The renin-angiotensin system is known for regulating blood pressure and body water balance as mediated by the octapeptide angiotensin II (AngII) acting at the angiotensin Type 1 receptor subtype (AT1; Figure 1). This system has received considerable attention with regard to the development of anti-hypertensive drugs. The RAS also promotes inflammation, oxidative stress, and tissue remodeling [57,58]), processes involved in what is now called the “neuronal inflammatory response” which is a key factor in neurodegenerative disease [59,60]. A contribution by AngII to memory formation was proposed some time ago [61]). Recent findings suggest that many of the memory enhancing effects originally attributed to AngII acting at the AT1 receptor subtype were due to the conversion of AngII to angiotensin III (AngIII) and then to the hexapeptide angiotensin IV (Ang IV) and it were AngIV activation of the AT4 receptor subtype that facilitated memory consolidation and retrieval [62-64]. This hypothesis is in agreement with the finding that angiotensin receptor blockers (ARBs) [65,66] and ACE inhibitors [67-69] improve cognitive processing in humans and animal models. Research interest in the AngIV/AT4 receptor system was further encouraged by the finding that AT1 and AT2 receptor antagonists failed to block learning and memory tasks in animal models [70]. The genesis of this prediction rested with the notion that AT1 receptor agonists should facilitate memory while antagonists were proposed to interfere with memory processing. In fact central blockade of AT1 receptors by ARBs facilitated a variety of memory tasks in rats and mice [48]; while central administration of AngII impaired spatial memory [71]. In contrast, a number of studies have noted AngIV and AngIV analog-induced facilitation of memory consolidation, retrieval and long-term potentiation (LTP), a phenomenon considered to be the building block of memory [48]. Taken together these findings encouraged research interest in understanding the role of the AngIV/ AT4 receptor system in cognitive facilitation and this system’s potential value as a treatment for AD.
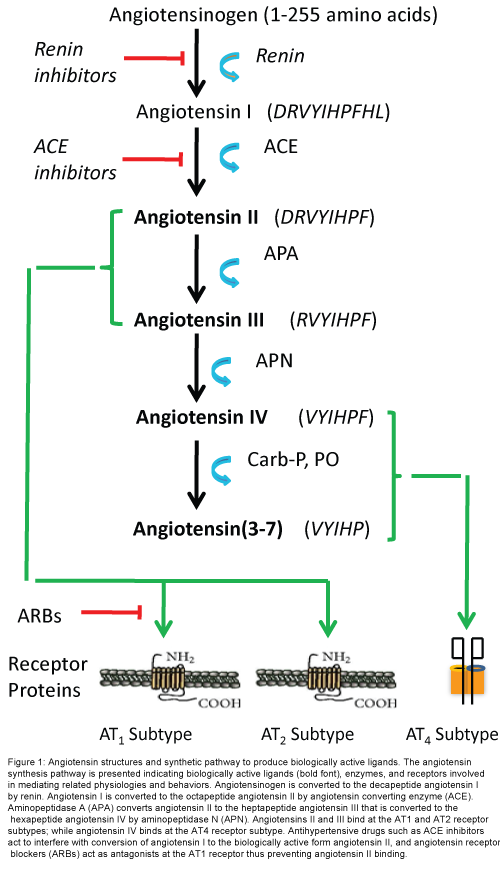
Figure 1: Angiotensin structures and synthetic pathway to produce biologically active ligands. The angiotensin synthesis pathway is presented indicating biologically active ligands (bold font), enzymes, and receptors involved in mediating related physiologies and behaviors. Angiotensinogen is converted to the decapeptide angiotensin I by renin. Angiotensin I is converted to the octapeptide angiotensin II by angiotensin converting enzyme (ACE). Aminopeptidase A (APA) converts angiotensin II to the heptapeptide angiotensin III that is converted to the hexapeptide angiotensin IV by aminopeptidase N (APN). Angiotensins II and III bind at the AT1 and AT2 receptor subtypes; while angiotensin IV binds at the AT4 receptor subtype. Antihypertensive drugs such as ACE inhibitors act to interfere with conversion of angiotensin I to the biologically active form angiotensin II, and angiotensin receptor blockers (ARBs) act as antagonists at the AT1 receptor thus preventing angiotensin II binding.
In the course of conducting research on this system we noticed a functional overlap between the AngIV/AT4 and HGF/Met receptor systems. These overlapping functions include facilitation of LTP and calcium signaling, memory enhancement, vascular angiogenesis, facilitation of blood flow and cerebroprotection, augmentation of dendritic arborization and synaptogenesis, and neurogenesis [48]. This observation led to the hypothesis that AngIV and AngIV analogs may be interacting with the HGF/Met system. This neurotrophic system is described in the next section (Figure 1).
The Hepatocyte Growth Factor/Met Receptor System
The plasminogen family member HGF, also known as “scatter factor”, acts at the Met receptor to stimulate mitogenesis, motogenesis and morphogenesis in a number of cellular targets including epithelial, endothelial and neurons [72-74]. This system has received considerable research attention related to its role in solid tumor cancers and possible therapies [75-77]. As the name implies HGF was originally isolated from the liver and shown to promote liver regeneration [78]. The Met receptor is made up of disulfide bond-linked alpha (45 kDa) and beta (145 kDa) subunits [79] (Figure 2). Met’s molecular weight agrees with our estimated weight for the AT4 receptor calculated some years ago and suggests that they are the same protein. The alphachain of Met is extracellular while the beta-chain is transmembrane. HGF dimerization precedes binding to the Met receptor which then undergoes phosphorylation. Once phosphorylated the tyrosine residues of the beta subunit serve as docking sites for downstream signaling mediators including extracellular signal-regulated kinase (ERK) and the phosphatidylinositol-3-kinase (P13K) pathway [78] (Figure 2).
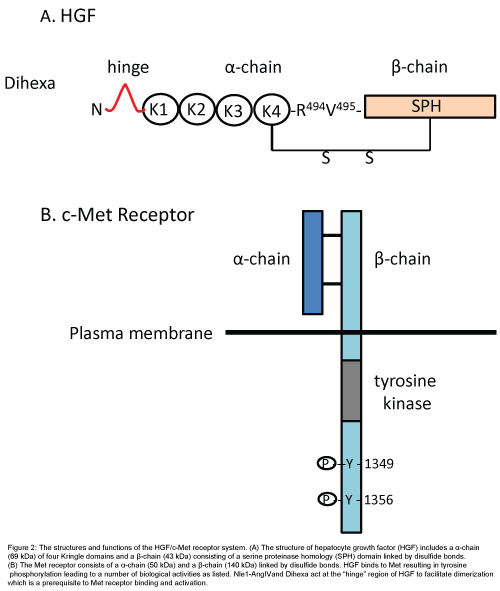
Figure 2: The structures and functions of the HGF/c-Met receptor system. (A) The structure of hepatocyte growth factor (HGF) includes a α-chain (69 kDa) of four Kringle domains and a β-chain (43 kDa) consisting of a serine proteinase homology (SPH) domain linked by disulfide bonds. (B) The Met receptor consists of a α-chain (50 kDa) and a β-chain (140 kDa) linked by disulfide bonds. HGF binds to Met resulting in tyrosine phosphorylation leading to a number of biological activities as listed. Nle1-AngIVand Dihexa act at the “hinge” region of HGF to facilitate dimerization which is a prerequisite to Met receptor binding and activation.
Given the overlapping functions listed above, members of our laboratory proposed that the AngIV/AT4 system closely interacts with the HGF/Met system [37,39,80,81]. This hypothesis was further supported by a search for a molecular target with a structure similar to AngIV. A partial match was seen with the protein angiostatin, and a related member of the plasminogen family HGF. This discovery suggested that AngIV-induced behaviors and physiologies could be mediated via the HGF/Met system. A major step toward understanding this relationship occurred when we determined that the AT4 receptor antagonist Norleual-AngIV (Nle-Tyr-Leu-(CH2-NH2)3-4-His-Pro-Phe) inhibited HGF binding to Met and HGF-dependent cell signaling, proliferation, invasion, and scattering [81]. We further determined that Norleual-AngIV’s mechanism of action as a Met antagonist is by blocking HGF dimerization which, as indicated above, is a prerequisite to binding and activation of the Met receptor [82,83]. This dimerization process is dependent upon a short HGF domain located between its N-terminal and first kringle domain referred to as the “hinge region” [83] (Figure 2). The importance of this hinge region was confirmed by the synthesis and utilization of a hexapeptide mimic (Hinge: KDYIRN) that bound to HGF with high affinity and blocked HGF dimerization [38]. The application of Hinge did not interfere with memory in normal functioning animals [84], a finding consistent with an earlier report that noted no impact on learning and memory in cognitively intact animals treated with AngIV and AngIV analogs [85]. Given these results we hypothesized that AngIV analogs mimic this hinge region and behave as allosteric activators by emulating the change in HGF’s conformation that normally results from its dimerization. Imaging data from cultured neonatal rat hippocampal neurons indicated that Nle1- AngIV (Nle-Tyr-Ile-His-Pro-Phe; Figure 3) stimulates dendritic spine numbers and size, as well as overall dendritic arborization, suggesting a plausible mechanism for enhanced synaptic plasticity, connectivity among neurons, and facilitation of memory [37]. HGF activation of the Met receptor has also been shown to mediate dendritic arborization and neurogenesis in cultured hippocampal neurons [86] and facilitate memory consolidation and retrieval in memory compromised animal models [87-90]. Elevated CNS levels of HGF have been measured in patients diagnosed with multiple sclerosis, Parkinson’s disease, amyotrophic lateral sclerosis and spinal cord injury [91-94]. Unfortunately these increases in HGF are not maintained with disease progression, and the hippocampal HGF/Met system appears to be down regulated in AD patients [95]. Thus, the brain HGF/Met system appears to initially respond to neurodegenerative disease-induced injury by facilitating synaptic plasticity and neurogenesis; however these elevations in HGF are not sustained as the disease progresses (Figure 3).
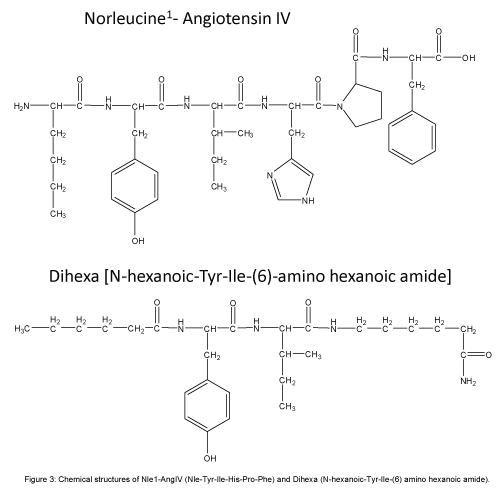
Figure 3: Chemical structures of Nle1-AngIV (Nle-Tyr-Ile-His-Pro-Phe) and Dihexa (N-hexanoic-Tyr-Ile-(6) amino hexanoic amide).
Design of Small Molecule AngIV Analogs
In order to further understand the AngIV/AT4 system’s role in learning and memory and the minimal structural requirements necessary for AngIV to facilitate memory consolidation we combined radio-receptor binding techniques with behavioral testing. We began by evaluating C-terminal shortened AngIV compounds for binding affinity. We learned from previous work conducted in our laboratory that the N-terminus of the molecule is critical for binding and activity [96-98]. Substitution of valine with norleucine in the first position enhanced activity [85]. Given this finding we began examining Nle1- AngIV and found that as C-terminal amino acids were removed Ki decreased from 3.59 × 10-12 for Nle1-AngIV to 4.89 × 10-9 M for Nle1- tripeptide (Nle-Tyr-Ile). Nle1-dipeptide (Nle-Tyr) had a Ki of 1.00 × 10-6 M indicating a substantial decrease in binding efficacy [99]. We then prepared rats each with an intracerebroventricular (ICV) guide cannula in order to deliver this compound directly into the brain given their resistance in crossing the BBB. Following recovery from surgery we behaviorally tested these animals utilizing the Morris water maze task of spatial learning which has become one of the “gold standard” behavioral testing protocols for the evaluation of AD animal models [37]. The icv delivery of the cholinergic muscarinic receptor antagonist scopolamine is frequently used to mimic the memory deficits seen in AD patients [100]. Those rats pretreated with scopolamine followed by Nle1-AngIV, Nle1-pentapeptide, or artificial cerebrospinal fluid (aCSF) followed by aCSF, all performed equivalently and evidenced effective search strategies. Those groups pretreated with scopolamine followed by Nle1-tetrapeptide, or Nle1-tripeptide, required longer durations to locate the platform, however their search patterns were effective. The dipeptide Nle-Tyr was ineffective in facilitating successful search strategies. We next synthesized small molecules that were metabolically stabile using methods to protect against peptidase action while maintaining biological activity. Four strategies were used to protect the N-terminal: 1) acylation of the N-terminal amine; 2) substitution of a d-amino acid at the N-terminal position; 3) substitution of a non- α-amino acid (e.g., β-amino acid or ?-amino acid) at the N-terminal; or 4) use of a peptide bond isostere in place of the 1-2 peptide bond. These peptides may also contain an amide at the C-terminal to reduce the action of carboxypeptidases. Most recently we have been successful in creating a tripeptide analog (eg. Dihexa) with enhanced hydrophobicity and reduced hydrogen bonding potential that is orally active and maintains agonist activity at the HGF/Met receptor (Figure 3). Despite these improvements imparted to Nle1-AngIV to construct pharmacokineticly superior Dihexa, Dihexa should not be considered the optimal drug candidate. Additional refinements to its structure to limit both phase 1 and phase 2 hepatic metabolism and first pass clearance, to reduce metabolism by gastric enzymes and enhance oral bioavailability, and to diminish polar surface area and overall size to facilitate BBB penetrability are currently in progress.
These results encouraged the possibility that a clinically useful drug can be designed possessing oral efficacy, BBB penetrability, increased metabolic stability, coupled with facilitated cognitive functioning. Subsequent efforts yielded a family of Dihexa-based molecules possessing increased hydrophobicity, decreased hydrogen bonding potential, and increased metabolic stability. Dihexa and its analogs bind with high affinity to HGF, induce Met phosphorylation in the presence of subthreshold levels of HGF, stimulate hippocampal spinogenesis and synaptogenesis equivalent with HGF [84], and promote neurogenesis and cerebroprotection (data in preparation for publication). Treatment with the HGF antagonist, Hinge as well as a short hairpin RNA directed at Met, significantly inhibited these processes. These compounds penetrate the BBB in sufficient quantity to facilitate memory consolidation and retrieval in aged rats, and the scopolamine-induced amnesic rat model of AD, as measured employing the Morris water maze task of spatial memory [80].
Therapeutic Prospective and Limitations
Limiting side effects is of particular importance regarding angiotensin-based antihypertensive drugs given the documented problems of dry mouth, nausea and dizziness, muscle soreness, and diuresis that may occur with ACE inhibitors and ARBs. Each member of these classes of drugs is designed to reduce AT1 receptor activation and control hypertension. However, the AngII/AT1 receptor system influences multiple functions beyond blood pressure, including body water balance, control of vasopressin and oxytocin release and sexual reproduction and behavior, thus undesirable drug-induced effects are possible. Dihexa-based compounds do not interact with central or peripheral AT1 receptors, are highly target specific, exhibit little interaction with cardiac channel proteins and hepatic CIP isoforms, and reveal no acute toxicity following a 6x effective dose of Dihexa. More extensive safety studies are currently underway. These data predict that the greatest clinical impact may be in individuals with moderately compromised brain HGF/Met systems as present in earlyto mid-stage AD. The combined neuroprotective, synaptogenic, and neurogenic mechanisms activated by these compounds encourage the possibility that they may offer a treatment option for neurodegenerative and neuro-traumatic disorders beyond AD. Despite the potential to attenuate and possibly reverse deleterious molecular events common to many neurodegenerative diseases, we do not foresee this approach as a “cure” because the underlying etiologies will likely continue, albeit at a slower rate. We do believe that this approach can attenuate damage due to ongoing neurodegenerative processes and thus sustain the patient’s quality of life for additional months of reduced symptomatology.
Conclusion
Nerve growth factor was the first neurotrophic agent to be discovered followed by BDNF [101]. Recently NT-3 and -4 have been isolated in the mammalian brain. HGF was originally extracted from liver and has now been identified in the brain [102,103]. Neurotrophins promote neural survival while substantial decreases in their levels have been measure in several neurodegenerative diseases. Thus, neurotrophins have been suggested as potential treatments for AD, Parkinson’s disease and other neurodegenerative diseases [104]. Recently we have designed and synthesized AngIV-based small molecules that influence HGF dimerization and in turn Met receptor activation. These compounds are orally active and penetrate the BBB at sufficient levels to improve cognitive functioning in animal models of AD. The therapeutic value of this approach lies in its capacity to encourage the formation of new functional synaptic connections among existing neurons, encourage the replacement of damaged and lost neurons from available neural stem cell populations, and facilitate cerebral blood flow and neuroprotection. Such treatment outcomes would benefit patients afflicted with AD and possibly those suffering from other neurodegenerative diseases.
Acknowledgements
Drs. Wright and Harding are co-founders of M3 Biotechnology, Inc. and hold stock in this company which is involved in the development of AD and Parkinson’s disease drugs. No funds from M3 were used in the writing of this manuscript. Although the authors have financial interest in this biotechnology company we acknowledge no conflict of interest in the preparation of this review article which represents an impartial and accurate presentation of research findings. All animal experiments conducted in our laboratory and presented in this manuscript adhered to the Guidelines for the Care and Use of Laboratory Animals as required by the National Institutes of Health (NIH Publication No. 80-23), and these protocols were approved by the Washington State University Institutional Animal Care and Use Committee.
9486
References
- Honig LS, Boyd CD (2013) Treatment of Alzheimer's Disease: Current Management and Experimental Therapeutics. CurrTranslGeriatrExpGerontol Rep 2: 174-181.
- Yaari R, Corey-Bloom J (2007) Alzheimer's disease. SeminNeurol 27: 32-41.
- Clark CM (2000) Clinical manifestations and diagnostic evaluation of patients with Alzheimer’s disease. In: Neurodegenerative Dementias: Clinical Features and Pathological Mechanisms, Clark, CM, Trajanowski JQ (Eds) McGraw-Hill, New York, pp 95-114.
- Maiese K, Chong ZZ, Hou J, Shang YC (2009) New strategies for Alzheimer's disease and cognitive impairment. Oxid Med Cell Longev 2: 279-289.
- Pouryamout L, Dams J, Wasem J, Dodel R, Neumann A (2012) Economic evaluation of treatment options in patients with Alzheimer's disease: a systematic review of cost-effectiveness analyses. Drugs 72: 789-802.
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, et al. (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9: 63-75.
- de la Torre JC (2009) Carotid artery ultrasound and echocardiography testing to lower the prevalence of Alzheimer's disease. J Stroke Cerebrovasc Dis 18: 319-328.
- Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184-185.
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356.
- de la Torre JC, Stefano GB (2000) Evidence that Alzheimer's disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev 34: 119-136.
- Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, et al. (2015) Vascular dysfunction in the pathogenesis of Alzheimer's disease--A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis 82: 593-606.
- de la Torre JC (2004) Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 3: 184-190.
- Kelleher RJ, Soiza RL (2013) Evidence of endothelial dysfunction in the development of Alzheimer's disease: Is Alzheimer's a vascular disorder? Am J Cardiovasc Dis 3: 197-226.
- van Norden AG, van Dijk EJ, de Laat KF, Scheltens P, Olderikkert MG, et al. (2012) Dementia: Alzheimer pathology and vascular factors: from mutually exclusive to interaction. BiochimBiophysActa 1822: 340-349.
- Farkas E, Luiten PGM, Bari F (2007) Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res Rev 54: 162-180.
- Kalaria RN, Harik SI (1989) Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53: 1083-1088.
- Yamagata K, Tagami M, Takenaga F, Yamori Y, Itoh S (2004) Hypoxia-induced changes in tight junction permeability of brain capillary endothelial cells are associated with IL-1beta and nitric oxide. Neurobiol Dis 17: 491-499.
- Cai Z, Zhao B, Ratka A (2011) Oxidative stress and β-amyloid protein in Alzheimer's disease. Neuromolecular Med 13: 223-250.
- Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178-201.
- Abbott NJ (2000) Inflammatory mediators and modulation of blood-brain barrier permeability. Cell MolNeurobiol 20: 131-147.
- Grammas P, Samany PG, Thirumangalakudi L (2006) Thrombin and inflammatory proteins are elevated in Alzheimer's disease microvessels: implications for disease pathogenesis. J Alzheimers Dis 9: 51-58.
- Grammas P (2011) Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflammation 8: 26.
- de la Torre JC, Aliev G (2005) Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab 25: 663-672.
- Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, et al. (2000) Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J DevNeurosci 18: 417-421.
- Walsh T, Donnelly T, Lyons D (2009) Impaired endothelial nitric oxide bioavailability: a common link between aging, hypertension, and atherogenesis? J Am GeriatrSoc 57: 140-145.
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. J NeuropatholExpNeurol 60: 759-767.
- Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, et al. (2006) Involvement of oxidative stress in Alzheimer disease. J NeuropatholExpNeurol 65: 631-641.
- de la Torre JC (2002) Alzheimer's disease: how does it start? J Alzheimers Dis 4: 497-512.
- de la Torre JC (2012) Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer's disease. J Alzheimers Dis 32: 553-567.
- Brayne C, Matthews FE, Xuereb JH (2001) Pathological correlates of late onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357: 169-175.
- Chen M, Maleski JJ, Sawmiller DR (2011) Scientific truth or false hope? Understanding Alzheimer's disease from an aging perspective. J Alzheimers Dis 24: 3-10.
- Ding Q, Dimayuga E, Keller JN (2006) Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal 8: 163-172.
- Polidori MC, Pientka L (2012) Bridging the pathophysiology of Alzheimer's disease with vascular pathology: the feed-back, the feed-forward, and oxidative stress. J Alzheimers Dis 28: 1-9.
- Boyce VS, Mendell LM (2014) Neurotrophic factors in spinal cord injury. HandbExpPharmacol 220: 443-460.
- Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. HandbExpPharmacol 220: 223-250.
- Williams AJ, Umemon H (2014) The best-laid plans go oft awry: Synaptogenic growth factor signaling in neuropsychiatric disease. Synaptic Neurosci 6: 1-20.
- Benoist CC, Wright JW, Zhu M, Appleyard SM, Wayman GA, et al. (2011) Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle1-angiotensin IV analogs. J PharmacolExpTher 339: 35-44.
- Kawas LH, Yamamoto BJ, Wright JW, Harding JW (2011) Mimics of the dimerization domain of hepatocyte growth factor exhibit anti-Met and anticancer activity. J PharmacolExpTher 339: 509-518.
- Kawas LH, McCoy AT, Yamamoto BJ, Wright JW, Harding JW (2012) Development of angiotensin IV analogs as hepatocyte growth factor/Met modifiers. J PharmacolExpTher 340: 539-548.
- Albiston AL, Diwakarla S, Fernando RN, Mountford SJ, Yeatman HR, et al. (2011) Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br J Pharmacol 164: 37-47.
- Wright JW, Kawas LH, Harding JW (2013) A Role for the Brain RAS in Alzheimer's and Parkinson's Diseases. Front Endocrinol (Lausanne) 4: 158.
- Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP, van den Bussche H (2005) Cholinesterase inhibitors for patients with Alzheimer's disease: systematic review of randomised clinical trials. BMJ 331: 321-327.
- Melnikova I (2007) Therapies for Alzheimer's disease. Nat Rev Drug Discov 6: 341-342.
- Thomas SJ, Grossberg GT (2009) Memantine: a review of studies into its safety and efficacy in treating Alzheimer's disease and other dementias. ClinInterv Aging 4: 367-377.
- Parsons CG, Danysz W, Dekundy A, Pulte I (2013) Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer's disease. Neurotox Res 24: 358-369.
- Wilkinson D (2012) A review of the effects of memantine on clinical progression in Alzheimer's disease. Int J Geriatr Psychiatry 27: 769-776.
- Galimberti D, Scarpini E (2011) Alzheimer's disease: from pathogenesis to disease-modifying approaches. CNS NeurolDisord Drug Targets 10: 163-174.
- Wright JW, Harding JW (2015) The Brain Hepatocyte Growth Factor/c-Met Receptor System: A New Target for the Treatment of Alzheimer's Disease. J Alzheimers Dis 45: 985-1000.
- Christensen DD (2006) Changing the course of Alzheimer’s disease: Emerging modifying therapies. Curr Psychiatry Rev 2: 179-188.
- Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ (2008) Amyloid-degrading enzymes as therapeutic targets in Alzheimer's disease. Curr Alzheimer Res 5: 212-224.
- Imbimbo BP, Giardina GA (2011) γ-secretase inhibitors and modulators for the treatment of Alzheimer's disease: disappointments and hopes. Curr Top Med Chem 11: 1555-1570.
- Saxena U (2010) Alzheimer's disease amyloid hypothesis at crossroads: where do we go from here? Expert OpinTher Targets 14: 1273-1277.
- Grammas P, Martinez J2, Sanchez A2, Yin X2, Riley J2, et al. (2014) A new paradigm for the treatment of Alzheimer's disease: targeting vascular activation. J Alzheimers Dis 40: 619-630.
- Schubert D, Maher P (2012) An alternative approach to drug discovery for Alzheimer's disease dementia. Future Med Chem 4: 1681-1688.
- Cook C, Stankowski JN1, Carlomagno Y1, Stetler C1, Petrucelli L1 (2014) Acetylation: a new key to unlock tau's role in neurodegeneration. Alzheimers Res Ther 6: 29.
- Frost B, Götz J, Feany MB (2015) Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol 25: 46-53.
- Dzau V, Braunwald E (1991) Resolved and unresolved issues in the prevention and treatment of coronary artery disease: a workshop consensus statement. Am Heart J 121: 1244-1263.
- Phillips MI, de Oliveira EM (2008) Brain renin angiotensin in disease. J Mol Med (Berl) 86: 715-722.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918-934.
- Ohshima K, Mogi M, Horiuchi M (2013) Therapeutic approach for neuronal disease by regulating renin-angiotensin system. CurrHypertens Rev 9: 99-107.
- Wright JW, Kawas LH, Harding JW (2015) The development of small molecule angiotensin IV analogs to treat Alzheimer's and Parkinson's diseases. ProgNeurobiol 125: 26-46.
- Braszko JJ, Walesiuk A, Wielgat P (2006) Cognitive effects attributed to angiotensin II may result from its conversion to angiotensin IV. J Renin Angiotensin Aldosterone Syst 7: 168-174.
- Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, et al. (2004) The angiotensin IV/AT4 receptor. Cell Mol Life Sci 61: 2728-2737.
- Wright JW, Clemens JA, Panetta JA, Smalstig EB, Weatherly LA, et al. (1996) Effects of LY231617 and angiotensin IV on ischemia-induced deficits in circular water maze and passive avoidance performance in rats. Brain Res 717: 1-11.
- Basso N, Paglia N, Stella I, de Cavanagh EM, Ferder L, et al. (2005) Protective effect of the inhibition of the renin-angiotensin system on aging. RegulPept 128: 247-252.
- Hajjar I, Keown M, Frost B (2005) Antihypertensive agents for aging patients who are at risk for cognitive dysfunction. CurrHypertens Rep 7: 466-473.
- Kehoe PG, Wilcock GK (2007) Is inhibition of the renin-angiotensin system a new treatment option for Alzheimer's disease? Lancet Neurol 6: 373-378.
- Ohrui T, Matsui T, Yamaya M, Arai H, Ebihara S, et al. (2004) Angiotensin-converting enzyme inhibitors and incidence of Alzheimer's disease in Japan. J Am GeriatrSoc 52: 649-650.
- Yamada K, Horita T, Takayama M, Takahashi S, Takaba K, et al. (2011) Effect of a centrally active angiotensin converting enzyme inhibitor, perindopril, on cognitive performance in chronic cerebral hypo-perfusion rats. Brain Res 1421: 110-120.
- Wright JW, Harding JW (2004) The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memory. ProgNeurobiol 72: 263-293.
- Tota S, Goel R, Pachauri SD, Rajasekar N, Najmi AK, et al. (2013) Effect of angiotensin II on spatial memory, cerebral blood flow, cholinergic neurotransmission, and brain derived neurotrophic factor in rats. Psychopharmacology (Berl) 226: 357-369.
- Bottaro DP, Rubin JD, Faletto DL, Chan AM, Kmiecik TE, et al. (1991) Identification of the hepatocyte growth factor receptor as the c-Met proto-oncogene product. Science 251: 802-804.
- Ma PC, Maulik G, Christensen J, Salgia R (2003) c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 22: 309-325.
- Skead G, Govender D (2015) Gene of the month: MET. J ClinPathol 68: 405-409.
- Awad AJ, Burns TC, Zhang Y, Abounader R (2014) Targeting MET for glioma therapy. Neurosurg Focus 37: E10.
- Benvenuti S, Comoglio PM (2007) The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol 213: 316-325.
- Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, et al. (2005) Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev OncolHematol 53: 35-69.
- Nakamura T, Mizuno S (2010) The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. ProcJpnAcadSer B PhysBiolSci 86: 588-610.
- Shinomiya N, VandeWoude GF (2003) Suppression of met expression: a possible cancer treatment. Commentary re: S. J. Kim et al., reduced c-Met expression by an adenovirus expressing a c-Met ribozyme inhibits tumorigenic growth and lymph node metastases of PC3-LN4 prostate tumor cells in an orthotopic nude mouse model. Clin. Cancer Res., 14: 5161-5170, 2003. Clin Cancer Res 9: 5085-5090.
- McCoy AT, Benoist CC, Wright JW, Kawas LH, Bule-Ghogare JM, et al. (2013) Evaluation of metabolically stabilized angiotensin IV analogs as procognitive/antidementia agents. J PharmacolExpTher 344: 141-154.
- Yamamoto BJ, Elias PD, Masino JA, Hudson BP, McCoy AT, et al. (2010) The angiotensin IV analog Nle-Tyr-Leu-(CH2-NH2)3-4-His-Pro-Phe (Norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J PharmacolExpTher 333: 161-173.
- Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, et al. (2006) Structural basis of hepatocyte growth factor/scatter factor and MET signalling. ProcNatlAcadSci U S A 103: 4046-4051.
- Youles M, Holmes O, Petoukhov MV, Nessen MA, Stivala S, et al. (2008) Engineering the NK1 fragment of hepatocyte growth factor/scatter factor as a MET receptor antagonist. J MolBiol 377: 616-622.
- Benoist CC, Kawas LH, Zhu M, Tyson KA, Stillmaker L, et al. (2014) Theprocognitive and synaptogenic effects of angiotensin IV-derived peptides are dependent on activation of the hepatocyte growth factor/c-met system. J PharmacolExpTher 351: 390-402.
- Wright JW, Stubley L, Pederson ES, Kramar EA, Hanesworth JW, et al. (1999) Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J Neurosci 19: 3952-3961.
- Tyndall SJ, Walikonis RS (2007) Signaling by hepatocyte growth factor in neurons is induced by pharmacological stimulation of synaptic activity. Synapse 61: 199-204.
- Date I, Takagi N, Takagi K, Kago T, Matsumoto K, et al. (2004) Hepatocyte growth factor attenuates cerebral ischemia-induced learning dysfunction. Biochem Biophysics Res Comm 319: 1152-1138.
- Date I, Takagi N, Takagi K, Kago T, Matsumoto K, et al. (2004) Hepatocyte growth factor improved learning and memory dysfunction of microsphere-embolized rats. J Neurosci Res 78: 442-453.
- Kato T, Funakoshi H, Kadoyama K, Noma S, Kanai M, et al. (2012) Hepatocyte growth factor overexpression in the nervous system enhances learning and memory performance in mice. J Neurosci Res 90: 1743-1755.
- Shimamura M, Sato N, Waguri S, Uchiyama Y, Hayashi T, et al. (2006) Gene transfer of hepatocyte growth factor gene improves learning and memory in the chronic stage of cerebral infarction. Hypertension 47: 742-751.
- Kato S, Funakoshi H, Nakamura T, Noma S, Kanai M, et al. (2003) Expression of hepatocyte growth factor and c-Met in the anterior horn cells of the spinal cord in the patients with amyotrophic lateral sclerosis (ALS): Immunohistochemical studies on sporadic ALS and familial ALS with superoxide dismutase 1 gene mutation. ActaNeurophathol 106: 112-120.
- Müller AM, Jun E, Conlon H, Sadiq SA (2012) Cerebrospinal hepatocyte growth factor levels correlate negatively with disease activity in multiple sclerosis. J Neuroimmunol 251: 80-86.
- Salehi Z, Rajaei F (2010) Expression of hepatocyte growth factor in the serum and cerebrospinal fluid of patients with Parkinson's disease. J ClinNeurosci 17: 1553-1556.
- Shimamura M, Sato N, Sata M, Wakayama K, Ogihara T, et al. (2007) Expression of hepatocyte growth factor and c-Met after spinal cord injury in rats. Brain Res 1151: 188-194.
- Hamasaki H, Honda H, Suzuki SO, Hokama M, Kiyohara Y, et al. (2014) Down-regulation of MET in hippocampal neurons of Alzheimer's disease brains. Neuropathology 34: 284-290.
- Krishnan R, Hanesworth JM, Wright JW, Harding JW (1999) Structure-binding studies of the adrenal AT4 receptor: Analysis of positions 2- and 3-modified angiotensin IV analogs. Peptides 20: 915-920.
- Sardinia MF, Hanesworth JM, Krebs LT, Harding JW (1993) AT4 receptor binding characteristics: D-amino acid- and glycine-substituted peptides. Peptides 14: 949-954.
- Sardinia MF, Hanesworth JM, Krishnan F, Harding JW (1994) AT4 receptor structure-binding relationship: N-terminal-modified angiotensin IV analogues. Peptides 15: 1399-1406.
- Wright JW, Harding JW (2009) The brain angiotensin IV/AT4 receptor system as a new target for the treatment of Alzheimer’s disease. Drug Develop Res 70: 472-480.
- Buccafusco JJ (2009) The Revival of Scopolamine Reversal for the Assessment of Cognition-Enhancing Drugs. The Revival of Scopolamine Reversal for the Assessment of Cognition-Enhancing Drugs.
- Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14: 7-23.
- Koike H, Ishida A, Shimamura M, Mizuno S, Nakamura T, et al. (2006) Prevention of onset of Parkinson's disease by in vivo gene transfer of human hepatocyte growth factor in rodent model: a model of gene therapy for Parkinson's disease. Gene Ther 13: 1639-1644.
- Martins GJ, Plachez C, Powell EM (2007) Loss of embryonic MET signaling alters profiles of hippocampal interneurons. DevNeurosci 29: 143-158.
- Woo KW, Kwon OW, Kim SY, Choi SZ, Son MW, et al. (2014) Phenolic derivatives from the rhizomes of Dioscoreanipponica and their anti-neuroinflammatory and neuroprotective activities. J Ethnopharmacology 155: 1164-1170.


