INTRODUCTION
|
| |
| The oral drug administration is by far the most preferable route for administration of drugs. However, the short biological half-life and preferential absorption via defined segment of intestine limits the therapeutic potential of many drugs[1], [2]. Such a pharmacokinetic limitation result in to frequent dosing to maintain the therapeutically effective blood concentration. This results in pill burden and consequently, patient complaints. The phenomenon of absorption via a limited part of the GIT has been termed the narrow absorption window; once this dosage form passes the absorption window, the drug will be neither bioavailable nor effective. In extreme cases drugs that are insufficiently absorbed due to narrow absorption window cannot be delivered entirely and are either given by the parenteral route or by the development of such dosage form, which is otherwise safe. |
| |
| Conventional oral dosage forms such as tablets, capsules provide specific drug concentration in systemic circulation without offering any control over drug delivery and also cause great fluctuations in plasma drug levels leading to peak plasma concentration at the absorption site and produce systemic toxicity and major draw-back is non-site specificity[3]. The drugs with site specific absorption, require drug release at specific site or such that maximum amount of drug reaches to the specific site. Pharmaceutical field is now focusing such drugs which require specificity. For oral solid-delivery systems, drug absorption is unsatisfactory and highly variable between the individuals despite excellent in vitro release patterns. There are several physiological difficulties such as inability to restrain and locate the controlled drug delivery system within the desired region of the gastrointestinal tract (GIT) due to variable gastric emptying and motility. Furthermore, the relatively brief gastric emptying time (GET) in humans which normally averages 2-3 h through the major absorption zone, i.e., stomach and upper part of the intestine can result in incomplete drug release from the drug delivery system leading to reduced efficacy of the administered dose. Therefore, control of placement of a drug delivery system in a specific region of the GI tract offers advantages for a variety of important drugs characterized by a narrow absorption window in the GIT or drugs with a stability problem[4], [5]. |
| |
| A rational approach to enhance bioavailability and improve pharmacokinetic and pharmacodynamic profile is to retain the drug reserve above its absorption region in GIT, i.e. in the stomach and to release the drug in controlled manner so as to achieve a zero order release kinetics (i.e. oral infusion) for prolonged period of time. |
| |
| Several attempts have been made to extend the gastrointestinal transit time of the dosage form. Sophisticated gastro retentive floating drug delivery systems are used for increasing gastric residence time. They remain in the gastric region for several hours and hence prolong the gastric residence time of the drug. It has several advantages over immediate release dosage form including the minimization of fluctuations in drug concentration in plasma and at the site of action over prolonged periods of time, resulting in optimized therapeutic efficiencies and reduce the side effects, reduction of total dose administered and reduction of administration frequency leading to improved patient compliance. |
| |
| Acyclovir, BCS class III/ IV drug [6], is widely used in the treatment of Herpes simplex virus infection as well as vericella zoster infection. It’s short biological half life ( 2.5-3.3 h) and low absorption (15-30% of administered dose) only from upper part of GIT are the two major reasons for the need of development of novel drug delivery system. Hence, it was aimed to develop gastro retentive system of acyclovir which results in to complete absorption and higher bioavailability. |
| |
| The present work consists of preparation and evaluation of floating microspheres of acyclovir using ethyl cellulose in different proportions. The drug is slowly released from microspheres and complete and slow drug release in the stomach is expected to increase bioavailability of the drug as well as its complete utilization which may result in to lower side effects. |
| |
MATERIALS AND METHODS
|
| |
| Acyclovir was obtained as gift sample from Bakul Pharma Pvt. Ltd (Ankleshwar, India). Ethyl cellulose was procured from S. D. Fine Chem Labs (Mumbai, India). Acetone and dichloromethane (DCM) were purchased from fine chemicals Ltd (Mumbai, India). All the chemicals used in the study were of analytical grade. |
| |
|
Preparation of floating microspheres
|
| |
| Acyclovir loaded microspheres were prepared by w/o/w emulsion solvent evaporation method[7]. In this method, acyclovir was dissolved in 10 ml 0.1N NaOH solution. The drug solution was then added to the solvent blend consisting of acetone: dichloromethane containing of ethyl cellulose while being stirred and then this solution was ultrasonicated for 15 min [8]. The water/oil emulsion was poured into aqueous phase containing 0.2 %v/v Tween 80 as stabilizer and gently allowed to stirr using a mechanical stirrer at high speed to obtain w/o/w emulsion. The emulsion was then brought to 40°C and stirred continuously for 3 h to evaporate the organic solvent blend. Microspheres were centrifuged at 1200 rpm for 20 minutes, washed twice with water, collected on a whatmann filter paper and dried under vacuum. |
| |
|
Design of experiments
|
| |
| A 32 full factorial was applied to design the experiments [9]. Polymer to drug ratio and stirring speed were used as independent variables, whereas % encapsulation efficiency, time required for 80% drug release, and particle size were kept as dependent variables. Formulations F1 to F9 were prepared using three different levels of polymer to drug ratio and stirring speed. Microspheres thus obtained were filtered, washed with water, and dried overnight at room temperature. The responses of the dependent variables were evaluated. The polynomial equations were generated for each responses using Design Expert Software (7.1.4). The check point batch was prepared to validate the polynomial equation. A 32 full factorial design layout is shown in Table 1. |
| |
|
Evaluation of formulations subjected to optimization
|
| |
|
Percentage drug encapsulation efficiency
|
| |
| The floating microspheres equivalent to 10 mg of acyclovir were accurately weighed and crushed. The powdered microspheres were dissolved in dichloromethane (5 ml) in volumetric flask and made the volume with 0.1 N HCl. This solution was then filtered through whatmann filter paper No. 44. After suitable dilution, the absorbance was measured at 254 nm using UV spectrophotometer and the percentage drug entrapped was calculated (equation 1). The drug entrapment study was conducted in triplicate. |
| |
 (1) (1) |
| |
|
Size and Surface morphology of the microspheres
|
| |
| The size distribution in terms of average diameter (davg) of the microspheres was determined by an optical microscopic method. A compound microscope fitted with a calibrated ocular diameter and stage micrometer slide was used to count at least 100 particles (Olympus, NWF 10x; Educational Scientific stores, India). The samples for the SEM analysis were prepared by sprinkling the microspheres on one side of a double adhesive stub. The stub was then coated with gold (Fine coat, ion sputter, JFC-1100). The microspheres were then observed with the scanning electron microscope at sophisticated instrumentation center for applied research and testing (SICART), Vallabh Vidyanagar, India using model (ESEM TMP+EDAX, Philips, Netherland) at 30 kv. |
| |
|
Micromeritic properties and % yield
|
| |
| The microspheres were characterized for true density, tapped density, Carr’s index (Ic), Hausner’s ratio (HR) and angle of repose using the following equations[10]. The tapping method was used to determine the tapped density and Carr’s index as follows. |
| |
| D0 = True density = W/V0 (2) |
| |
| Dt = tapped density =W/Vt (3) |
| |
| Hausner’s ratio (HR) = Tapped density/True density (4) |
| |
| Carr’s index = (Tapped density – True density)/Tapped density *100 (5) |
| |
| V0 and Vt are the true volume and tapped volume respectively. |
| |
|
In vitro evaluation of floating ability
|
| |
| The floating microspheres (300 mg) were spread over the surface of the dissolution medium 0.1N HCl, pH (1.2) containing 0.02%v/v of Tween 80 that was agitated by a paddle rotated at 100 rpm. After agitation for a predetermined time interval, the microspheres that floated over the surface of the medium and those settled at the bottom of the flask were recovered separately. After drying, each fraction of the microspheres was weighed and their buoyancy was calculated by the following equation: |
| |
| Buoyancy (%) = Qf / Qf + Qs (6) |
| |
| Qf = Quantity of floating microspheres, Qs = Quantity of settled microspheres |
| |
|
In vitro drug release study
|
| |
| The in-vitro release of acyclovir from the ethyl cellulose microspheres was measured in 0.1N HCl at 37°C±0.5°C using USP paddle dissolution apparatus type II (LAB INDIA DISSO 2000). An accurately weighed amount of prepared microspheres equivalent to 100 mg of pure Acyclovir, were stirred in 900 ml dissolution medium at 100 rpm. Samples were withdrawn at predetermined time interval and replaced immediately with same volume of fresh medium. Aliquots following suitable dilution were analyzed spectrophotometrically at 254 nm. The drug release experiments were conducted in triplicate following the above procedure. |
| |
|
Drug- Excipients compatibility study Fourier transform infrared spectroscopy
|
| |
| Infrared spectra of acyclovir, ethyl cellulose blank microspheres and acyclovir loaded microspheres were taken by using KBr pellet technique and were recorded on a Fourier transform Infrared spectrophotometer (FTIR-8400S, Shimadzu, japan). |
| |
|
Differential Scanning Calorimetry
|
| |
| Calorimetric analysis was performed at S. K. Patel Pharmacy College, Kherva, India using model DSC-&, Perkin Elmer, equipped with a measuring cell DSC 20. About 2 mg sample was placed in pierced aluminium pans and heated at a scanning rate of 10ºC per minute from 50 to 250 ºC. The instrument was calibrated with an indium standard. |
| |
|
Data analysis
|
| |
| A statistical model incorporating interactive terms was used to evaluate the responses: |
| |
| Y= β0 + β1X1 + β2X2 + β12X1X2 (7) |
| |
| Where β0, the intercept is the arithmetic average of all quantities outcomes of nine runs, β1, β2 and β12 are the coefficients computed from the observed experimental values of Y, and X1 and X2 are the coded levels of the independent variables. The terms X1X2 (i = 1, 2) is the interaction terms. |
| |
|
Stability studies
|
| |
| The stability studies were carried out at an optimized formulation, i.e., formulation F9. The formulation was stored at 40º ± 2ºC/75% ± 5% RH for 3 months[12], [13]. After intervals of 7, 15, 30, 60, and 90 days, samples were withdrawn and retested for drug release. Paired t-test was used to compare the dissolution profiles. |
| |
RESULTS AND DISCUSSION
|
| |
|
Percentage drug encapsulation efficiency
|
| |
| The percentage encapsulation efficiency of microspheres varied from 59%-77%w/w (Table 2). Results demonstrated that an increase in polymer to drug ratio, increased the encapsulation efficiency of the drug. The drug encapsulation efficiency was good because the drug was sparingly soluble in the aqueous medium. Stirring speed had a negative effect on entrapment efficiency. |
| |
|
Size and surface morphology of microspheres by Scanning electron microscopy
|
| |
| The SEM micrographs revealed that the resulting microspheres were spherical in nature with rough surfaces containing cracks and holes over its surface. The micrographs showed almost spherical but the morphology appeared to be rough (Figure 1). The reason behind this morphology change can be attributed to the faster evaporation of dichloromethane forming a pore like structure. The mean particle size of the microspheres significantly increased with increase in polymer concentration and was in the range of 275-340 µm (Table 2). |
| |
|
Micromeritic properties and % yield
|
| |
| The compressibility index ranged from 13.70% to 16.75% (Table 2). All formulations showed excellent flowability as expressed in terms of angle of repose in the range of 17º-26º (Table 2). The percentage yield of prepared microspheres was in the range of 76%- 95% (Table 2). |
| |
|
In vitro evaluation of floating ability
|
| |
| Floating ability of batch F9 microspheres was found to be more than about 12 h due to insolubility of the ethyl cellulose polymer in 0.1N HCl (pH 1.2). The results also showed a tendency that the larger the particle size, the longer the floating time. It should be noted, however, that the situation in vivo can be quite different and the residence time may vary widely depending on the phase of gastric motility. |
| |
|
In vitro drug release study
|
| |
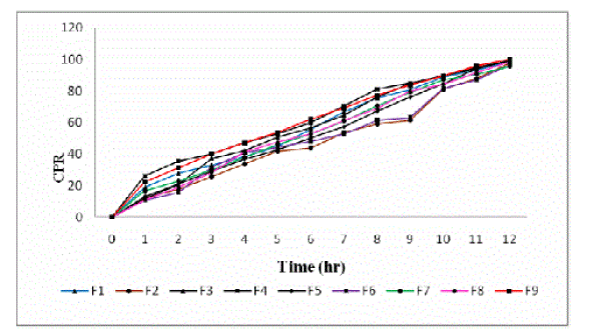
| The in vitro drug release profiles of all the formulations have been shown in figure 2. The release of acyclovir mainly depends upon the polymer concentration. The release rate of the drug from the microspheres was found to decrease drastically with increase in polymer concentration. Acyclovir release from all the formulations was found to be slow and sustained over 12 h. Bye the end of 12 h, Formulations F1, F2, F3, F4, F5, F6, F7, F8 and F9 were found to release 98.69%, 97.47%, 98.86%, 99.28%, 99.17%, 96.46%, 95.65%, 98.27% and 99.97% respectively (Figure 2). |
| |
|
Data Analysis
|
| |
| The floating microspheres of acyclovir were prepared by varying polymer to drug ratio (X1) and stirring speed (X2) as independent variables. The time required for 80% drug release (T80% Y1), percentage encapsulation efficiency (Y2) and particle size (Y3) were taken as dependent variables. On the basis of the data obtained from the formulations subjected to optimization, a general statistical model can be depicted with respect to the above data. The model developed can be characterized by using the polynomial equation representing the respective response data. This can be given as follows: In order to make prediction of Y1, Y2 and Y3 mathematical models were evolved omitting the insignificant terms. Equations 1, 2 and 3 represents the refined models for responses Y1, Y2 , and Y3 with values of R2 and Fischer’s ratio (F). |
| |
| Y1= 519.11+ 29.66X1 - 1.66X2 (R2= 0.996, F=13.71, P<0.05) (8) |
| |
| Y2= 64.92 + 6.01X1 + 0.92X2 (R2 = 0.946 F= 24.79, P<0.05) (9) |
| |
| Y3= 287.89 + 20.50X1 +1.0X2 + 10.50X1X2 (R2 = 0.986 F= 27.75, P<0.05) (10) |
| |
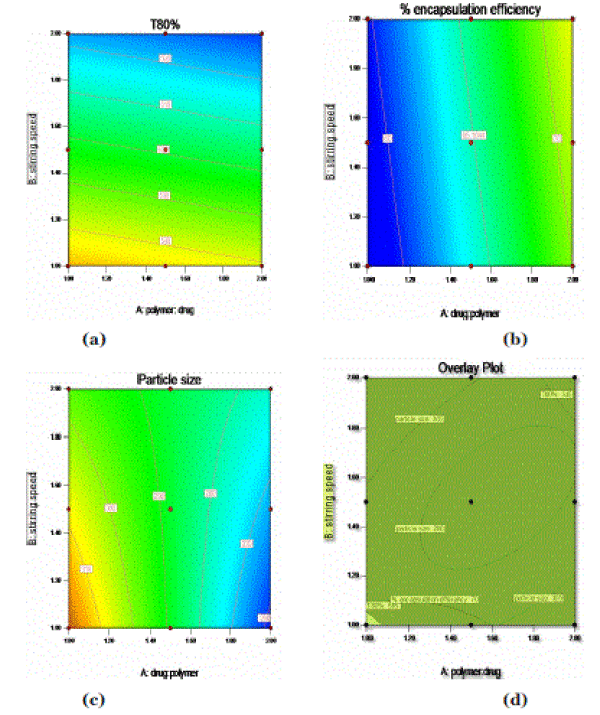
| From the above equations, contour plots of the respective responses were generated, which were then used to predict the responses of dependent variables at the intermediate levels of independent variables. In the case of Y1 (T80%), coefficients (β1= 29.6) and (β2= 1.66) were found to be significant. As the polymer to drug ratio (X1) was increased, the T80% was increases (Figure: 3(a)). In case of response Y2, coefficients (β1= 6.01) and (β2= 0.92) were found to be significant (Figure: 3(b)). As the polymer to drug ratio (X1) was increased, the percentage encapsulation efficiency was increased. In case of response Y3, coefficients (β1= 20.50) and (β2= 1.0) were found to be significant. As the polymer to drug ratio was increased, the particle size was decreased (Figure: 3(c)). |
| |
|
Validation of optimum floating microspheres formulation
|
| |
| Table 5 lists the composition of the checkpoints, shows good correlation plots between the observed and the predicted values of drug entrapment efficiency, T80%, and particle size which confirms the practicability of the model. |
| |
|
Model fitting kinetic
|
| |
| The in vitro release data of batch F10 were analyzed for establishing kinetics of drug release. Model fitting was done using an in-house program FORTRAN. Zero order, First order, Higuchi[11], Hixson-Crowell, Korsemeyer-peppas and Weibull models were tested. The best fit was shown by Korsemeyer- Peppas model with least sum of square of residuals (SSR) and Fischer’s ratio (Table 3). |
| |
|
Drug- Excipients compatibility study
|
| |
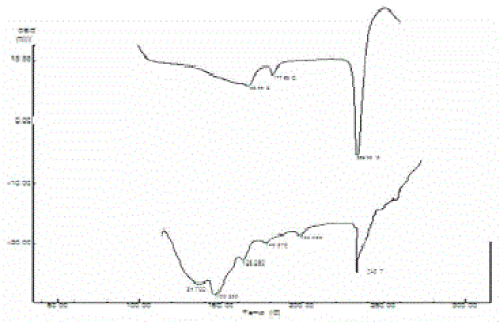
| Pure acyclovir showed prominent peaks at 3458 cm-1, 1731 cm-1 and 3029 cm-1 because of hydroxyl, carboxy and aromatic amine respectively. These peaks were retained in acyclovir loaded microspheres indicating the stability of acyclovir during processing of microspheres (Figure 4). This was further supported by DSC. Acyclovir showed a sharp endothermic peak that corresponds to melting in the range of 250-300 ºC, as shown in Figure 4(a). Acyclovir in the ethyl cellulose microspheres Figure 4(b) also showed a similar characteristic peak with decreased intensity showing its stability during the encapsulation. |
| |
|
Stability studies
|
| |
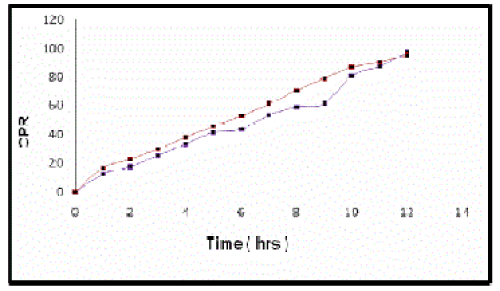
| Stability studies indicated that there was no significant difference observed between the release pattern of microspheres at 40ºC and 75%RH and at room temperature for three months[12],[13] (Figure 5). |
| |
CONCLUSIONS
|
| |
| The results of a 32 full factorial design revealed that the polymer to drug ratio (X1) and stirring speed (X2) significantly affected the dependent variables such as drug encapsulation efficiency, T80% and particle size of the microspheres. The best fiited model was Korsemeyer-peppas. The microspheres of the check point batch (F10) exhibited 70.76% drug encapsulation efficiency, mean particle size of 310 µm and 570 min T80% (Table 3) which were nearer to predicted values obtained from overlay contour plot of all the responses, (Figure 3(d)). An appropriate balance between the levels of the polymer to drug ratio and stirring speed was imperative to acquire maximum drug encapsulation efficiency, sustained release of the drug and adequate particle size. Hence, it could be established that among the prepared formulations, F9 was the optimum formulation. In vitro data obtained for the floating microspheres of acyclovir showed excellent floating ability, good buoyancy and prolonged drug release. |
| |
ACKNOWLEDGEMENTS
|
| |
| We are thankful to Gujarat Council of Science and Technology (GUJCOST) for providing financial assistance to carry out research work. We are grateful to Bakul Pharma Pvt. Ltd, Loba Cheme Pvt. Ltd, S D fine chemicals Ltd. (Mumbai, India) for providing us gift samples. |
| |
Conflict of Interest
|
| |
| NIL |
| |
Source of Support
|
| |
| NONE |
| |
Tables at a glance
|
 |
|
|
|
| Table 1 |
Table 2 |
Table 3 |
Table 4 |
|
| |
Figures at a glance
|
 |
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
Figure 5 |
|
| |


