Introduction
The currently accepted explanation of the short action of barbiturate anesthetics is that the redistribution of drug within the body compartments and also some degree of metabolism being involved. It is, however manifestly evident that the barbiturates anesthetic are not rapidly metabolized. Thus it was assumed that molecular modifications could provide barbiturates which undergo rapid metabolism and thereby would offer very important clinical advantage. It was felt that there was a need for an agent that is potent, very brief because of rapid metabolic break down and allows restoration of all mental faculties within 1-2 hours without persistent hangover or headache.
Despite isolated attempts to inject drugs into the veins, the procedure remained a desperate remedy for emergencies only. It was not until Paul Ehrlich [1] used the intravenous injection of arsphenamine that the technique came into vogue. Further, the widespread administration of intravenous saline during the World War-II finally ensured the universal administration of the technique in clinical practice.
The first drug used as intravenous anesthetics was hexobarbitone, but this was quickly replaced by shorter acting drug, thiopentone. But due to its very short duration of action its use remained limited to simple surgical procedure lasting only for a few minutes. Besides, thiopentone tends to accumulate in the body after repeated administration, its use was limited to that of induction agent only and in addition considerable cautions had to be excesised by the anesthetists in view of its low therapeutic index.
The activity of barbiturates may be greatly modified by altering their lipid solubility. This may be achieved either by introducing suitable groups in side chain or alkylation, arylation of ring nitrogen as well as substitution of sulfur and selenium for the ring oxygen. Investigations by Mautner (1970) revealed that by increasing the side chain length up to 8 carbon atoms, both lipophilicity and potency rise but beyond 8 carbon atoms produces convulsions or inactive compounds [2]. A probable explanation is that the lipophilicity has reached at such a level that the drug in question is unable to reach bio-phases. On the other hand the introduction of alkyl or aryl in the ring nitrogen often results in lipid solubility which increases in anesthetic activity with shorter duration of action as seen with thiopentone with sulfur in 2-position than pentobarbitone. Thus, highly lipid soluble anesthetic barbiturates equilibrate very rapidly between plasma and brain tissues resulting anesthesia within a few seconds. Sulfur and Selenium in 2-positions of barbiturates increase acidity and thereby have greater ability to withdraw electron from -NH group [3-5].
The metabolic conversion of barbiturates is believed to play an important role in the early fall of their plasma concentration. The most important route of metabolism appears to be its side chain oxidation and N-dealkylation or desulfuration, as appropriate. The metabolites resulting from the oxidative process may be a ketone, an aldehyde or an acid depending on the nature of the drug. Thus, the metabolism on oxidation results an inactive species. On the other hand N-de-alkylation it gives active metabolites. For example, mephobarbitone on de-alkylation produces active barbiturate phenobarbitone. It has also been seen with N-dealkylation, desulfuration results in the retention of pharmacological activity in the metabolic products.
It is well known that the presence of an ester function in many cholinergic, anti-cholinergic and local anesthetic agents is not only responsible for binding with receptor at the site of action, but is also responsible for the susceptibility of these drugs to the action of plasma esters and as such these compounds are rapidly metabolized. Again, the introduction of ester functions into the decamethylene chain of the neuromuscular blocking agent (NBA) decamethonium has been exploited to overcome the prolonged action of this compound. The resulting drug suxamethonium is being widely used in surgery.
The currently accepted explanation of the short action of barbiturate anesthetics could be that the redistribution of drug within the body compartments and some degree of metabolism also being involved. It is, however manifestly evident that the barbiturates anesthetic are not rapidly metabolized. It has been assumed that molecular modifications could provide barbiturates which undergo rapid metabolism and thereby would offer very important clinical advantage. We feel that there is a need for an agent that is potent, very brief because of rapid metabolic break down and allows restoration of all mental faculties within 1-2 hours without persistent hangover or headache. It is the aim of this research described here to meet these requirements of rapid metabolism in the particular case of anesthetic barbiturates.
In view of the above, it was felt that by incorporating a metabolically labile ester function in the barbiturate side chain, it might be possible to dsign a rapidly metabolized anesthetic agent. The barbiturate esters reported in the first series of this work has proved marked improvement in both their potency, duration of action and faster recovery than with thiopentone.
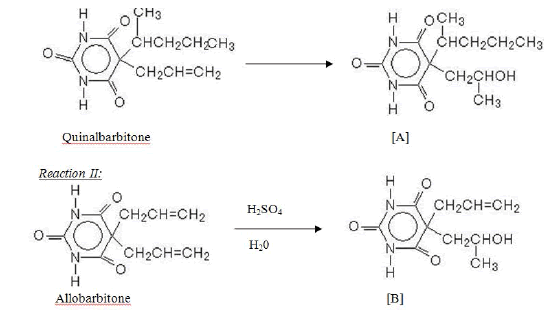
The present investigation began with a systematic search of literature for a convenient route for the synthesis of barbiturate esters. It was found that 5-alkyl-5-allyl barbiturate could be converted to 5-alkyl-5-(2-hydroxypropyl)-barbiturate (1). Thus for preliminary investigation and pharmacological studies we selected quinalbarbitone (5-[(2R)-pentan-2-yl[-5-prop-2enyl-1,3- diazinane-2,4,6-trione) and allobarbitone (5,5-diprop-2-enyl-1,3- diazinane-2,4,6-trione) and converted into 5-(1-methylbutyl)- 5-(2-hydroxypropyl) barbituric acid [A] and 5-allyl-5-(2- hydroxypropyl) barbituric acid [B] respectively.
Experimental
These hydroxylated barbituric acids were esterified with different alkyl groups confirmed their structure by elemental analysis and pharmacological screening were carried out following:
Melting points measurement: Melting points were recorded by Reichert’s microscopic melting point apparatus.
TLC: Silica gel plates both extemporaneously prepared plates (MN-Duren, MN-Kieselgel G-HR) and (MN-Duren polygram sil G/ UV 254) plates were found to be satisfactory.
Development solvent: A mixture of chloroform: acetone in the ratio 1:1
Spray reagent: Saturated aqueous solution of silver acetate; Diphenylcarbazone in 10% aqueous ethanol.
Experimental Procedure: The plate on spraying produces a lilac spot on the chromatogram. The barbiturate esters were tested in male mice (CFLP) usually in groups of 10(18-20 gm). Drugs were administered intravenously, as sodium salts in physiological saline; m, w, c indicates 5% mulgofen dissolved in CO2 free distilled water. Percentage death figure in the table refers to acute toxicity, whilst delayed toxicity (24 hours) mentioned in brackets. Certain esters administered in Dutch albino rabbits weighing 1-2 kg. Elemental analyses were done in the Department of Chemistry, Strathclyde University, George Street, Glasgow, UK. Pharmacological studies were carried out by Mr. Neil Duff of Aspro-Nicolas Laboratories, Slough, England.
Synthesis of 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid [A]: 25 g of Quinal albarbitone was taken in a round bottomed flask and 25 ml of conc. sulfuric was added to it at room temperature (25°C) and stirred constantly for 30 minutes. The deep red colored solution was put into a conical flask containing 200 ml iced water. On keeping for about 30 minutes in acidic water crystalline solid appeared. The solid crystals were filtered, washed with iced cold water and dried the in air for 2-3 hours. The air dried product was then dried in an oven at 60-70°C for 2 hours and purified.
Purification: The product was dissolved in boiling water and activated charcoal (5-7%) was added to the hot solution, stirred further for 10 minutes and after cooling at room temperature, crystals of the product appeared. This was filtered through filter paper, washed with hot water and dried in the oven in around 80°C temperature until free from moisture. White crystals (20 g, 74.5%) of 5-(1-methylbutyl)-(2-hydroxypropyl) Barbituric acid (mp. 191-193C, Lit. 204) were obtained
Analysis Found: C-56.60; H-7.90; N-10.85; O-24.65%. C12H20N2O4 requires: C-56.0; H-7. 90; N-10.90 and O-25.2%: Rf. 0.50
Synthesis of 5-Allyl-5-(2-hydroxypropyl) barbituric acid [B]:10 g of Allobarbitone was treated with 20 ml of conc. sulfuric acid for 5 minutes at room temperature (25°C). After pouring this solution into 100 ml of iced cooled water and keeping it for 8 hours a crystalline product was obtained. The product was filtered, washed, dried and re-crystallized from ether (commercial) and dried as in A to get colorless crystals of 5-allyl-5-(2-hydroxypropyl) barbituric acid, mp 167-169°C.Yield: 5.8 g (72.5%).
Analysis Found: C-53.0; H-5.90; N-12.10, and O-29.0%. C10H14N4 requires: C-53.0; H-6.20; N-12.40%; and O-28.4%. Rf. 0.43
Synthesis of esters from compound (a):
(I) 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid acetate (I): 2 g of 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid was refluxed in 6 ml acetic anhydride in a round bottomed flask for 6 hours. The reaction mass was cooled (room temp.) and iced water added to it when colorless solid appeared. The solid was filtered, washed with water and dried in an oven at 75- 80°C for 2 hours. The dried product was dissolved in ether and chromatographed over basic alumina in a column (400 mm X 20 mm) when the ethereal elute on removal of solvent gave colorless oil. The oil was dissolved in petroleum ether (40-60) when the needle shaped crystals appeared. The product was filtered and dried in an oven at 100oC for 2 hours (until free from moisture) to get 1.73 g of the ester (73%), mp 168-170oC.
Analysis Found: C-56.60; H-7.70; N-9.50% ; and O-26.2%: C14H22N2O5 requires: C-56.40; H- 7.40; N- 9.40 and O-26.8%. Rf. 0.69
(II) 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid propionate: 2 g of [A] was refluxed with 10 ml of propionic anhydride for six (6) hours. The propionic ester was separated as in (I). The crude ester was purified in pet-ether (40-60) to get 1.82 g (74.5%, mp. 116-118°C).
Analysis Found: C-57.70; H-7.50; N-9.0 and O-25.80%. C15H24N2O5 requires: C-56.75; H-6.75; N-9.50 and O-27.0 %. Rf. 0.76
(III) 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid butyrate: 2 g of [A] was dissolved in 10 ml of butyric acid and 4-5 drops of conc. sulfuric acid added to it. The mixture was refluxed for 6 hours, cooled at room temperature and iced water was added. Extraction of aqueous mixture with ether and removal of the solvent gave an oily substance, which was dissolved in ether and dried over anhydrous magnesium sulfate. The solvent was removed and subjected to column chromatography on basic alumina and the ether eluant produced oil again. This was crystallized from ether-pet-ether (40-60; 1:1) to provide fine needles of 5-(1-methylbutyl)-5-(2-hydroxypropyl) barbituric acid butyrate (1.80 g, 71.0%), mp. 150-152°C.
Analysis Found: C-59.0; H-7.90; N-8.25 and O-24.85%.C16H26N2O5 requires: C-58.90; H-8.0; N-8.60 and O-24.50%. Rf.0.77
synthesis of esters from compound (b):
(IV) 5-Allyl-5-(2-hydroxypropyl) Barbituric acid acetate: 2 g of 5-allyl-5-(2-hydroxypropyl) barbituric acid [B] was dissolved in 6 ml of acetic anhydride and refluxed for 6 hours. After pouring on to iced water and leaving it for 8 hours a solid mass was appeared. The solid was separated by filtration washed properly with water and dried in an oven at 90°C for 5 hours. This was then re-crystallized from boiling water to give fine needles of 5-allyl- 5-(2-hydroxypropyl) barbituric acid acetate (1.70 g, 71.5%), mp. 141-142°C.
Analysis Found: C-54.0; H-6.0; N-10.40 and O-29.60%. C12H16N2O5 requires: C-53.70; H- 6.0; N-10.4 and O-29.90%; Rf. 0.66
(V) 5-Allyl-5-(2-hydroxypropyl) barbituric acid propionate: 2 g of product [B] and 6 ml of propionic anhydride were refluxed for 6 hours. On cooling an oil was deposited which was extracted with ether (15 ml), washed with water, dried over anhydrous magnesium sulfate and the solvent removed. The oil was subjected to column chromatography on basic alumina and the ether eluate on removal of solvent gave oil again. The oil was crystallized from ether-pet-ether (40-60; 1:1) which yielded fine needles of 5-allyl-5-(2-hydroxypropyl) barbituric acid propionate (1.66 g; 66.0%), mp.120-121°C.
Analysis Found: C-56.60; H-6.30; N-9.80 and O-27.30%: C13H18N2O5 Requires: C-55.30; H-6.40; N-9.80 and O-28.50%; Rf. 0.73
(VI) 5-Allyl-5-(2-hydroxypropyl) Barbituric acid butyrate: 2 g of [B] was dissolved in 6 ml of butyric anhydride in which 5-6 drops conc. sulfuric acid was added and the mixture refluxed for 6 hours. Following the procedure as in (V) fine needles of 5-allyl- 5-(2-hydroxypropyl) barbituric acid butyrate (1.60g; 61.0%), mp.130-131°C.
Analysis Found: C-56.60; H-6.85; N-9.40 and O-27.15%: C14H20N2O5 Requires: C-56.75; H-6.75; N-9.50 and O-27.0%; Rf. 0.76
Result and Discussions
In the first series (A), six barbiturate esters were prepared and administered in mice. Pentobarbitone and thiopentone were used as reference samples. It was observed that with compounds II and III recovery was markedly faster than with thiopentone, whilst the characteristic CNS excitation prior to anesthesia, as seen with pentobarbitone, did not occur in compound II and III. The rate of onset of anesthesia was rapid in compounds II and III compared to pentobarbitone and thiopentone. The selection of esters and 5-alkyl substitutions were taken in view of the work of Hansch relating to the potency of hypnotic barbiturates to their Octanol-water partition coefficients.
Conclusions
In view of the work, it was felt that by incorporating a metabolically labile ester function in the barbiturate side chain, it might be possible to design a rapidly metabolized anesthetic agent. The preliminary pharmacological results reported here support this view.
Acknowledgements
We are thankful to Mr. Neil Duff, Aspro-Nicolas Laboratories, and Slough, England for their help to carried out the studies also to the authority of Primeasia University for their fruitful encouragements to publish the research work. Authors also thankful to the commonwealth scholarship authority for their funding to carry on the research work.
6484
References
- Mautner HG, Clemson HC (1970) Medicinal Chemistry Ed. A Burger (3rd edtn), Willey Interscience, New York, London.
- Hill SA (2002) Correct nomenclature for stereo isomers. Anaesthesia 57: 200.
- Drayer DE (1986) Pharmacodynamic and pharmacokinetic differences between drug enantiomers in humans: an overview. Clin Pharmacol Ther 40: 125-133.


