Keywords
|
| Primary CNS lymphoma; Primary intra-ocular lymphoma; Uveitis |
Case Presentation
|
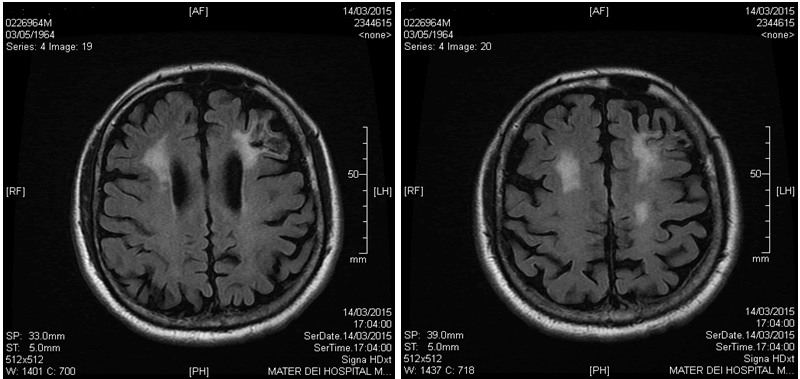
| A 50 year old man presented with an 18 month history of recurrent bilateral uveitis and reduced visual acuity. This was treated with topical steroids. One year later he presented with an acute confusional episode that resolved over a two week period. An MRI scan (Figure 1a and 1b) showed a diffuse infiltrative process, the differential diagnosis of which was inflammation, demyelination, sarcoidosis or lymphoma. He was treated with methylprednisolone for a presumptive diagnosis of sarcoidosis. The CT scan showed small volume mediastinal lymphadenopathy. The blood count and bone marrow studies were normal and there was no monoclonal immunoglobulin. The CSF was normal. |
| Because of a recurrence of the uveitis, he underwent a subretinal aspiration. The vitreous humour was infiltrated by lymphoplasmacytoid cells which on flow cytometry were confirmed to be monoclonal and of B- cell origin. They were positive for CD 19, CD 22, CD 79a and CD 38. They were also kappa light chain restricted. The diagnosis of a primary intraocular lymphoma (of low grade histology) was made on a background of primary CNS lymphoma. Treatment was initiated according to the Ferreri regimen with high dose methotrexate/cytarabine. He developed neurological toxicity during the second cycle leaving him with permanent neurological deficits. The treatment was subsequently attenuated to high dose methotrexate alone for four cycles. At the end of his therapy there was resolution of the cerebral lesions and the vitreous humour was acellular. |
Discussion
|
| Primary CNS lymphoma (PCNSL) is an aggressive form of non-Hodgkin’s lymphoma arising in and confined to the brain, spinal cord, leptomeninges, retina vitreous humour and occasionally optic nerve [1]. The ocular manifestation of PCNSL is termed primary intra-ocular lymphoma (PIOL) and that can occur prior to, concurrent with or subsequent to PCNSL. Although rare, the incidence of both PCNSL and PIOL seems to be increasing in the immunocompetent population. PCNSL represents 4% of intracranial neoplasms and 4-6% of all extranodal lymphomas. |
| PCNSL usually presents with neurological symptoms. A survey of 245 patients with PCNSL showed that 70% had focal neurological defects, 33% raised intracranial pressure, 14% seizures and 4% ocular symptoms [2]. Headache is a rare complaint. Up to 20% of patients with PCNSL present with ocular involvement which can masquerade as steroid resistant uveitis. This is usually bilateral and is often associated with painless loss of vision and/or vitreous “floaters” [3]. |
| Radiology usually reveals a mass lesion which is multifocal in over a third of cases. Frequently, PCNSL is not suspected on the basis of its radiological appearance which may mimic other CNS tumours or conditions like toxoplasma, sarcoidosis, demyelinating disorders or multifocal leucoencephalopathy [4]. |
| Diagnosis of PCNSL requires morphology and immune histochemical studies. Tissue is preferentially acquired through stereotactic surgery. In 90% of cases, PCNSL is subtyped as diffuse large B- cell lymphoma . The remainder are a mixture of Burkitt’s lymphoma, T- cell rich B- cell lymphoma, peripheral T- cell lymphoma and rarely low grade B cell non-Hodgkin’s lymphoma [5,6]. |
| Should the ocular manifestations of PCNSL occur prior to detection of cerebral involvement, the diagnosis is achieved by performing a vitreous biopsy. The vitrectomy specimens are notoriously acellular so much so that diagnostic failure rates of up to 30% have been reported. |
| PCNSL is highly sensitive to steroid therapy. Radiologically, resolution of disease is detectable within 48 hours in up to 40% of patients (vanishing tumour). This response is short lived with disease recurrence occurring soon after steroid withdrawal [7]. A response to steroids is not diagnostic of PCNSL as similar improvement can be seen in patients with conditions like sarcoidosis or multiple sclerosis. Steroid treatment should if possible be avoided before tissue biopsy as the treatment interferes with the histopathological picture. |
| The prognosis of PCNSL/PIOL is poor. Untreated, there is a median survival of six weeks for the high grade variety. For decades, radiotherapy was the exclusive treatment giving a median survival of 12 months, but the addition of cytotoxic drugs that cross the blood brain barrier has significantly improved the outcome. Methotrexate- based regimens can achieve a median survival of 60 months [8] and the high dose methotrexate/cytarabine combination is the current standard approach as it is supported by the highest level of evidence. However, neurological toxicity associated with long term therapy remains a problem. |
| The eye is a reservoir for PCNSL tumour cells. Systemic administration of high dose methotrexate/cytarabine yield therapeutic levels in the intraocular fluids and clinical responses have been documented. However drug concentrations in vitreous humour are unpredictable and intraocular relapse is common [9] For these reasons, patients with PCNSL with intraocular disease are treated with high dose methotrexate- based chemotherapy followed by whole brain radiotherapy and bilateral ocular irradiation which results in better disease control [10]. |
Key Points
|
| • Intraocular lymphoma (IOCL) is a variant of Primary CNS lymphoma (PCNSL). |
| • PCNSL/IOCL is a rare tumour, the incidence of which though is increasing. |
| • PCNSL/IOCL is difficult to diagnose and is usually diagnosed late. Subretinal aspiration is the less invasive than sterotactic surgery and has a reasonably good diagnostic yield. |
| • PCNSL/IOCL can mimic a variety of other conditions including uveitis. |
| • Institution of steroid therapy can dramatically interfere with the diagnostic process. |
| • There is general lack of awareness among physicians of its myriad deceptive presentations. |
| • The prognosis is poor, however there have been significant improvements in outcomes following the introduction of CNS penetrating chemotherapy regimens. |
| • Early diagnosis improves the chances of successful treatment. |
| • The success of treatment can be tempered by the significant neurotoxicity associated with the more intensive regimens. |
Figures at a glance
|
 |
| Figure 1 |
| |
|
|
|

