Ira Shukla* and Suneetha V
VIT University, Vellore, Tamil Nadu, India
Corresponding Author:
Ira Shukla
VIT University, Vellore-632 014, Tamil Nadu, India E-mail: irashukla1994@gmail.com
Received date: June 23, 2016; Accepted date: June 30, 2016; Published date: July 06, 2016
Citation: Shukla I, Suneetha V (2016) Tyrosine Kinase Inhibitor Therapy: Resistance and Mutation Detection in Chronic Myeloid Leukemia Patients. Int J Drug Dev & Res 8: 009-011
Keywords
Chronic myeloid leukemia; Imatinib resistance; Mutation; BCR-ABL
Introduction
At the molecular level, chronic myeloid leukemia is caused by the expression of bcr-abl protein which results from the expression of Philadelphia Chromosome [Ph]. This Ph chromosome containing the chimeric oncogene BCR-ABL is only found in patients whose chromosome 9 and 22 have fused due to the incidence of a reciprocal translocation. This gene expresses a protein tyrosine kinase which causes acceleration of cell division and restriction of DNA repair (Figure 1).
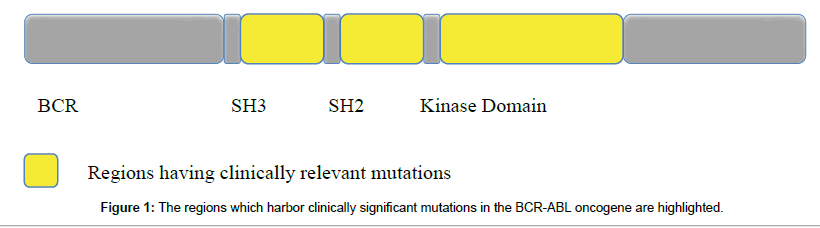
Figure 1: The regions which harbor clinically significant mutations in the BCR-ABL oncogene are highlighted.
CML disease progression is divided into Chronic Phase [CP], Accelerated Phase [AP] and culminates in Blast Phase [BP]. In CP cells grow exponentially but retain the capacity to function well. In AP, the immature blood cells increase in number losing the ability to function well and fight infections while in the BP the disease progresses to ALL.
First Line Treatment and Resistance
In order to curb the activity of bcr-abl tyrosine kinase, imatinib mesylate [STI571] was identified as a tyrosine kinase inhibitor. It replaced Interferon alpha as the first line of therapy and achieved durable results. Survival of up to 85% of the patients on Imatinib mesylate was confirmed by the International Randomized Study of Interferon versus STI571 [IRIS] trial while disease free progression was reported in 92% of the patients. The bcr-abl protein kinase is constitutively active. It binds to ATP and tyrosine residues on various substrates receive phosphate from it. This results in increased proliferation of cells. Imatinib is effective as it prevents ATP to bind to BCR-ABL tyrosine kinase arresting its activity. As the subsequent phosphorylation procedure stops, the adverse cellular events are terminated too.
Despite this progress up to 27% of patients who reach Complete Cytogenic Response and a few patients in the advanced phases of the disease show relapse or resistance while undergoing imatinib therapy [1]. Intrinsic resistance is also known as primary resistance and is caused in the absence of initial efficacy of drug. Acquired response or relapse is also known as secondary resistance and is caused when the drug loses its initial efficacy on the patient [2]. When BCR ABL1/ABL1 transcript levels are more than 10% in a patient or if he does not show Partial CyR [presence of less than equal to 35% Ph+ metaphase] after treatment for 3 months or does not exhibit Complete CyR [absence of Ph+ metaphases] after 12 or 18 months from start of treatment then the patient is exhibiting resistance according to the National Comprehensive Cancer Network guidelines.
Various mechanisms may be the explanation for this phenomenon. These include mutations in the binding site of the drug, bcr-abl gene may undergo amplification causing a hike in the amount of mRNA expressed [3], novel genes may be expressed which may lead to multi drug resistance, SRC, LYN or other oncogenes may also start getting expressed, Oct-1 which is a transported for the drug may experience reduction in its level of expression [4], BIM which induces apoptosis generates poor response to TKIs due to deletion type polymorphism.
Distribution of Mutations
The main regions where mutations are reported to occur and lead to drug resistance are P loop, A Loop, C Loop and the direct binding site [5]. The P Loop refers to the phosphate-binding loop. When imatinib binds, this loop undergoes a change in its conformation to allow a better fit for the drug so that it may easily associate with tyrosine 253 [Y353] with a hydrogen bond. Point mutations at Y253 will interfere with this binding. The A Loop refers to the Activation loop [6,7]. It closes the kinase active site making ABL inactive and thereby allowing the drug to bind. Thus mutations in this loop will hamper with ABL specificity and promote drug resistance. C Loop refers to the catalytic loop. M351T is an example of a mutation that occurs in this loop and causes changing the conformation of the ABL. The direct binding site refers to T315 which is responsible for formation of Hydrogen bond between the drug and ABL. A point mutation of threonine to isoleucine results in allosteric effects which disallow the drug to bind with ABL region and thus produces the strongest resistance to the TKIs. Mutations are also known to occur in SH2 and SH3 contact domains which denote the contact area for SH2 and SH3 domain containing proteins. They are situated outside the kinase domain in the linker kinase domain and auto inhibits the kinase domain. Mutations in these regions are known to case activation of the kinase [8-10] (Table 1).
| Structural motif |
Common sites for mutations |
| P Loop |
M244 |
| G250 |
| Q252 |
| Y253 |
| E255 |
| L248 |
| K247 |
| A Loop |
V379 |
| A380 |
| F382 |
| L384 |
| L387 |
| M388 |
| Y393 |
| H396 |
| A397 |
| C Loop |
M351 |
| E355 |
| Direct Binding Site |
T315 |
| F317 |
| F359 |
| SH3 Contact |
E292 |
| I293 |
| L298 |
| V299 |
| SH2 Contact |
Y342 |
| M343 |
| A344 |
| A350 |
Table 1: Shows common mutations found in the kinase domain (P loop, A Loop, C Loop, Direct Binding Site) and regulatory domain (SH3 contact, SH2 contact) of BCR-ABL gene which leads to resistance in patients.
Second Line Treatment
To counter the resistance caused by imatinib resistance, second generation TKIs were developed which were nilotinib, dasatinib, bosutinib and bafetinib. These, however were still ineffective against T315I mutation as a resultant of which third generation drug namely Ponatinib was developed [unavailable in India]. As each drug has a different efficacy for particular mutations it is vital to identify the type of mutation present in the patient’s ABI kinase domain so that the most effective drug for that particular mutation may be availed by the patient [11,12].
Nilotinib is a structurally modified version of imatinib which allowed it to be 30 times more effective than the former in terms of its anti-CML activity. Dasatinib has lower specificity than nilotinib even though its binding action to ABL is 325 times stronger as it suppresses an excess of 50 kinases including SRC family kinases [13]. It’s effective on more mutants than those covered by nilotinib but still has limited efficacy over patients with T315I, F317L and V299L. Bosutinib has a 50 times stronger activity than imatinib and causes lesser side effects due to its weak inhibition of c-KIT and PDGFR. Bafetinib is 55 times stronger than imatinib and can suppress all forms of mutations except T315I. It is also shown to be effective in patients suffering from Parkinson’s disease [14].
T315I is a vital mutation as even though it was the first mutation to be discovered, until recently there was no drug to treat it. Ponatinib is a third generation drug which has a proven efficacy against T315I. It counters the conformational effect caused by the mutation due the presence of a long carbon-carbon triple bond that gives it flexibility. PF- 114 is an oral drug which is known to have inhibited all mutations and suppressed the emergence of new mutations though clinical trials have yet to start. It is known to be more selective in action than dasatinib and ponatinib, Omacetaxicine can be used to treat patients with T315I mutations if they can not be treated with ponatinib [15].
Techniques for Mutation Analysis
On incidence of spike in BCR-ABL transcript levels, absence or inadequacy of response to drug, it is recommended to go for mutation analysis in the BCR-ABL kinase domain in order to aid better therapeutic course of action.
European Leukemia Net recommends Direct Sequencing procedure to detect mutations. Combining it with D-HPLC allows prescreening the samples for presence of variations and modifies the detection limit to 1% (22) [16]. The direct sequencing method using Sanger’s technique is known to have 15-25% sensitivity. Thus it’s not the ideal method for finding low level mutations. Using PCR the first amplification occurs of BCR exon 2 to ABL exon 10. In the nested PCR round ABL 4 to 10 exons are amplified. To verify the accuracy of the amplification, an aliquot is run on the gel to confirm the presence of bands ~863 base pairs which is followed by direct sequencing using Big Dye V3.1 chemistry.
Using Pyrosequencing we can achieve sensitivity up to 5% along with the added capacity to detect multi allelic mutations in mixed cell populations. It’s advantageous over other techniques due to its high specificity and in built control system [17]. However we can only use it if know beforehand which mutation we are looking for. After obtaining the target PCR product [by subjecting it to nested PCR as explained in direct sequencing] it is then subjected to pyrosequencing and quantification for each of the 6 relevant mutations takes place.
D-HPLC or WAVE refers to denaturing ion pair reverse phase liquid chromatography works on the principle of differentially retaining DNA which are homo or hetero duplex inside a matrix which separates the fragments on the basis of charge density in respect to electrolyte gradient [18]. It’s advantageous over others as it allows screening of unknown mutations at reduced expense with an excellent sensitivity [1%] [6]. Due to its operational complexity, and additional efforts required to interpret data as it fails to characterize mutations, it is not as ideal as the other techniques in a clinical setting. In a study conducted by Ernst et al. in 2009 it was found that though D-HPLC was able to detect at least 9 more mutations when combined with direct sequencing as opposed to performing direct sequencing alone, these extra mutations were only minor sub clones and hence this extra sensitivity was irrelevant statistically.
Novel methods which promise higher sensitivities in the detection of mutations include ultra-deep sequencing, mass spectrometry and digital PCR though none of them have been incorporated into routine clinical practice yet. The digital PCR helps in identifying kinetics of mutation. New generation sequencing or ultra-deep sequencing is advantageous over other methods because of its increased sensitivity [1-5%] for detecting any known and unknown mutations in regions other than the hotspots. If the BCR-ABL fusion transcript level is lower than 10% then direct sequencing method will miss most mutations. In this respect NGS is highly recommended as it can detect emerging or residual mutations and thus is useful for aiding therapy [19]. Sub cloning and sequencing has its own disadvantages as cloning takes time and is prone to contamination. High resolution melt curve analysis is sensitive to quality of DNA and gives false negative tests. Restriction fragment length polymorphism based assays have limited applicability and give rise to many false positives and false negatives. Allele specific PCR based genotyping method is laborious and gives low level mutations with unidentified clinical significance. Double gradient denaturing electrophoresis is extremely sensitive for a clinical setting with a large number of samples.
Conclusion
Thus we conclude that in order to provide targeted therapy for the patients who suffer from mutations and show absence of or under satisfactory response to first line therapy or exhibit cytogenic and hematological resistance in second line therapy, identification of mutation present in the individual is necessary using affordable and reproducible techniques. Sanger’s sequencing may not have a high sensitivity but is able to detect the major mutations with known clinical significance and is recommended for periodical analysis for presence of mutations. It may be used in conjugation with screening methods like D HPLC.
9939
References
- Serpa M, Sanabani SS, Dorliac-Llacer PE, Conchon M, Pereira TD, et al. (2010) Molecular measurement of BCR-ABL transcript variations in chronic myeloid leukemia patients in cytogenetic remission. BMC Blood Disord 10: 7.
- Hochhaus A (2006) Chronic myelogenous leukemia (CML): resistance to tyrosine kinase inhibitors. Ann Oncol 17: x274-x279.
- Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, et al. (2006) Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci USA 103: 19466-19471.
- Sherbenou DW, Hantschel O, Kaupe I, Willis S, Bumm T, et al. (2010) BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib. Blood 116: 3278-3285.
- Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, et al. (2005) Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood 105: 2093-2098.
- Alikian M, Gerrard G, Subramanian PG, Mudge K, Foskett P, et al. (2012) BCR-ABL1 kinase domain mutations: methodology and clinical evaluation. Am J Hematol 87: 298-304.
- Sherbenou DW, Wong MJ, Humayun A, McGreevey LS, Harrell P, et al. (2007) Mutations of the BCR-ABL-kinase domain occur in a minority of patients with stable complete cytogenetic response to imatinib. Leukemia 21: 489-493.
- Soverini S, Branford S, Nicolini FE, Talpaz M, Deininger MW, et al. (2014) Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leukemia 38: 10-20.
- Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, et al. (2014) BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 26: 428-442.
- Iqbal Z, Aleem A, Iqbal M, Naqvi MI, Gill A, et al. (2013) Sensitive detection of pre-existing BCR-ABL kinase domain mutations in CD34+ cells of newly diagnosed chronic-phase chronic myeloid leukemia patients is associated with imatinib resistance: implications in the post-imatinib era. PLoS One 8: e55717.
- Kimura S, Ando T, Kojima K (2014) BCR-ABL Point Mutations and TKI Treatment in CML Patients. J Hematol Transfus 2: 1022.
- Larson RA, Hochhaus A, Hughes TP, Clark RE, Etienne G, et al. (2012) Nilotinib vs. imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia 26: 2197-2203.
- Azam M, Latek RR, Daley GQ (2003) Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112: 831-843.
- Barila D, Superti-Furga G (1998) An intramolecular SH3-domain interaction regulates c-Abl activity. Nat Genet 18: 280-282.
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, et al. (2000) Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289: 1938-1942.
- Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, et al. (2006) Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res 12: 7374-7379.
- Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, et al. (2011) BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118: 1208-1215.
- de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, et al. (1982) A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 300: 765-767.
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, et al. (1984) Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 36: 93-99.


