Introducción
Las enfermedades reumáticas son causa de una gran demanda en los servicios de salud, se ha estimado que alrededor del 33% de la población general, en algún momento de su vida presenta una enfermedad reumática [1].
La AR, es una de las tres enfermedades más frecuentes en la consulta externa de Reumatología con una prevalencia estimada hasta del 47.1% [2].
Esta enfermedad, presenta un evidente riesgo de deterioro funcional, ya que se comprobó que una década posterior al inicio de los síntomas, al menos 50% de los pacientes son incapaces de mantener un trabajo de tiempo completo [3]. Además, los pacientes con AR presentan un incremento en la tasa de mortalidad [4].
La etiología de la AR permanece desconocida. Al igual que otras enfermedades autoinmunes, se caracteriza por una alteración en la respuesta inmune con presencia de inflamación crónica y producción de autoanticuerpos (Figura 1).
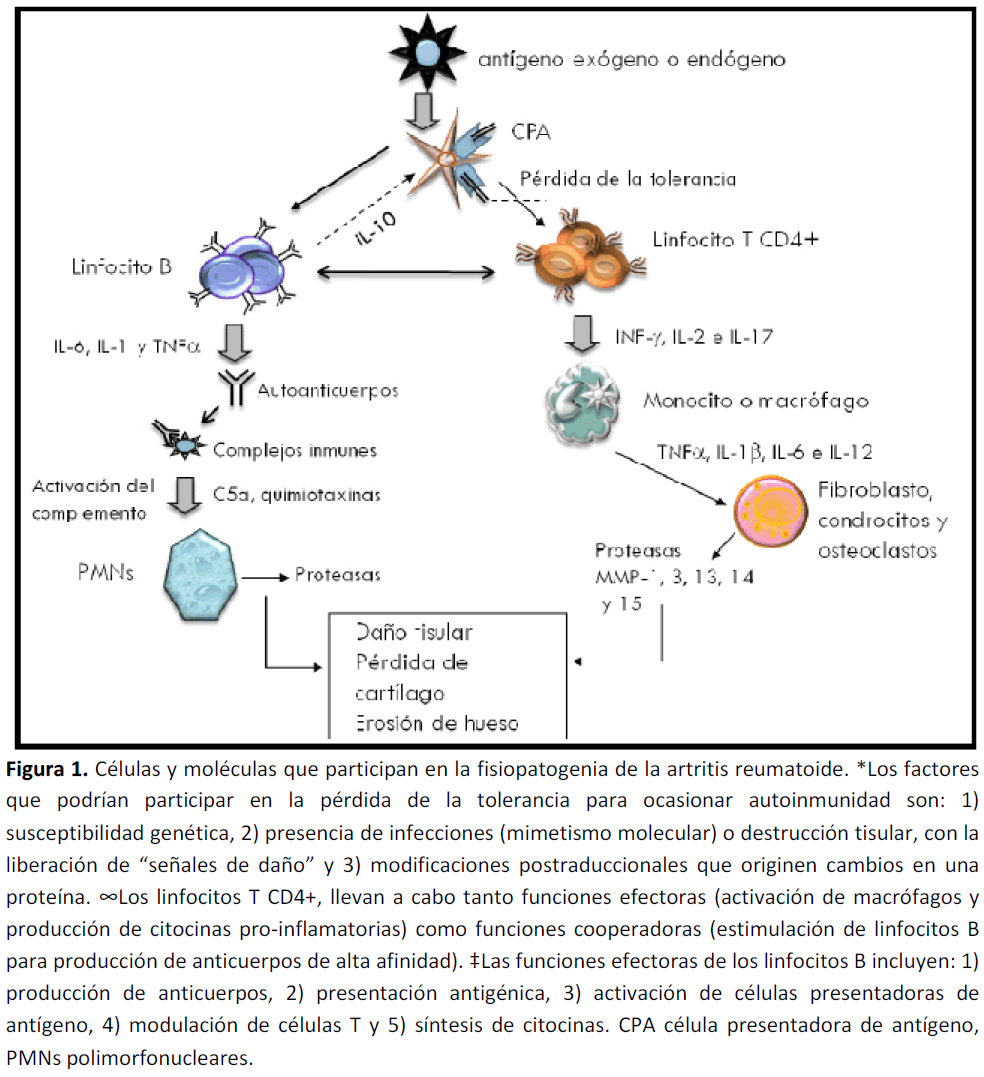
Figura 1. Células y moléculas que participan en la fisiopatogenia de la artritis reumatoide. *Los factores que podrían participar en la pérdida de la tolerancia para ocasionar autoinmunidad son: 1) susceptibilidad genética, 2) presencia de infecciones (mimetismo molecular) o destrucción tisular, con la liberación de “señales de daño” y 3) modificaciones postraduccionales que originen cambios en una proteína. ∞Los linfocitos T CD4+, llevan a cabo tanto funciones efectoras (activación de macrófagos y producción de citocinas pro-inflamatorias) como funciones cooperadoras (estimulación de linfocitos B para producción de anticuerpos de alta afinidad). ‡Las funciones efectoras de los linfocitos B incluyen: 1) producción de anticuerpos, 2) presentación antigénica, 3) activación de células presentadoras de antígeno, 4) modulación de células T y 5) síntesis de citocinas. CPA célula presentadora de antígeno, PMNs polimorfonucleares.
A través de los años, se han roto diversos paradigmas en cuanto a la patogénesis de esta enfermedad. En 1997 Weyand y Goronzy [5], propusieron un nuevo modelo hipotético para la fisiopatogenia de la AR, el cual integra factores de riesgo genéticos y la complejidad de las respuestas inflamatorias.
Este modelo asume que la respuesta inmune del huésped no está involucrada en el proceso inicial de la enfermedad, sino que en una primera etapa, el huésped se ve expuesto a un gran número de antígenos secundario a daño en tejido sinovial, por múltiples causas y que en forma paralela, varios genes de la respuesta inmune como los del HLA, genes de inmunoglobulinas y receptores de células T, tienen un impacto en el tipo de respuesta que se desarrolla contra estos antígenos. En una segunda etapa entonces, se origina la respuesta autoinmune, en donde las células del sistema inmune, específicamente linfocitos T auto-reactivos, montan una repuesta contra éstos antígenos y se origina todo un proceso inflamatorio alrededor, con la presencia de células como macrófagos, células T, B y neutrófilos, infiltradas en la membrana sinovial inflamada. La producción de anticuerpos y formación de complejos inmunes, que a su vez ocasionan el reclutamiento de una mayor cantidad de células inflamatorias y sus productos [6].
Durante la última década, la investigación sobre la patogénesis de la AR se ha centrado en la idea de que existe la participación de múltiples células y mecanismos efectores moleculares del sistema inmune. Sin embargo, de esta complicada vista emerge un punto en común, éste es, el escape al control inmunológico. De esta manera los aspectos fundamentales en la patogénesis de la enfermedad son el entendimiento de la pérdida de tolerancia, la activación de linfocitos T auto-reactivos y la generación de autoanticuerpos.
Hla: El Epítopo Compartido
Diversos estudios han demostrado ya, una fuerte correlación entre la presencia de AR y algunos alelos del HLA-DR. Los alelos HLA-DRB1*0401 y HLA-DRB1*0404 (que codifican para la molécula DR4dw4) son los más comúnmente encontrados y su presencia se asocia con una enfermedad más severa y con alta mortalidad [7,8]. Estas moléculas del HLA-DR, comparten una secuencia común en la tercera región hipervariable (epítopo compartido), éstos motivos consisten de los aminoácidos 70QKRAA74 y 70QRRAA74 en DRB1*0401 y DRB1*0404 respectivamente [9]. Estos cinco aminoácidos se encuentran en el sitio de unión al péptido y por lo tanto son de suma importancia durante la presentación antigénica.
Estudios genéticos, que evalúan la contribución del HLA a la fisiopatogenia de AR, han evidenciado que el riesgo a padecer esta enfermedad en hermanos de individuos afectados es 2-17 veces más alto que en los hermanos de individuos no afectados [10]. También se ha descrito la asociación entre AR y el complejo HLA en diferentes poblaciones, contándose aproximadamente hasta un tercio de componente genético para susceptibilidad en la enfermedad [10].
Autoanticuerpos en la Fisiopatogenia de la ar
Una característica prominente de la AR es la presencia de varios tipos de autoanticuerpos. Además, esta enfermedad es una de las pocas en las que la presencia de estructuras similares a centros germinales ectópicos, pueden observase en la membrana sinovial inflamada [11,12]. Esta característica sugiere la presencia, activación, diferenciación y producción local de anticuerpos por células B [13].
La contribución de estos anticuerpos a la patología inicia con la unión directa de dichos anticuerpos a sus antígenos, la formación de complejos inmunes, depósito y activación del complemento y receptores Fc, producción de factores quimiotácticos y reclutamiento de polimorfonucleares que participan en la reacción inflamatoria mediante la producción de proteasas que ocasionan daño tisular [14,15].
El anticuerpo mejor conocido y utilizado es el Factor Reumatoide (FR), el cual es un autoanticuerpo dirigido contra la porción Fc de las moléculas IgG. Aunque éste no es específico de la enfermedad se utiliza rutinariamente para la clasificación de AR [16].
Recientemente surgió interés en una nueva clase de autoanticuerpos específicos para la AR, los anticuerpos contra péptido citrulinado cíclico (aPCC), los cuales alcanzan una especificidad del 98% y sensibilidad de 80% [17,18]. Estos anticuerpos aPCC, pertenecen a la familia de autoanticuerpos contra el factor perinuclear (APF) [18,19]. El epítope de este grupo de anticuerpos son los residuos en los que la arginina es convertida por peptidil arginina desaminasa (PAD) a un producto citrulinado mediante modificaciones postraduccionales [20,21]. De ahí que a este grupo de autoanticuerpos se les conozca como anticuerpos contra proteínas citrulinadas.
Un estudio en particular, destaca la presencia de estos anticuerpos en individuos asintomáticos (donadores de sangre) que subsecuentemente desarrollarían AR; de esta manera, se les atribuye un alto valor predictivo positivo [22]. Esto indica que la citrulinización de proteínas y producción de anticuerpos, son procesos tempranos implicados en el desarrollo de AR [23,24].
Por otro lado, la presencia de proteínas citrulinadas en sinovio no se asocia a la presencia de aPCC en pacientes sin AR, ya que se demostró que aún cuando existen proteínas citrulinadas en sinovio inflamado por diversas causas, distintas de AR, no se detectó la presencia de anticuerpos aPCC [25]. Por este motivo, se ha postulado que la presencia de dichas proteínas con péptidos citrulinados pueden ser un fenómeno asociado a la inflamación [26], esto explicaría la presencia de citrulinación en artropatías inflamatorias diferentes a la AR.
En un estudio realizado por Verport y cols [27], se demostró la presencia de aPCC de clase IgM, de forma continua, durante el curso de la AR, esto indica que durante la enfermedad existe un reclutamiento activo de nuevas células B a la respuesta inmune originalmente montada; reflejando así una continua re-activación y la persistencia del antígeno desencadenante de la producción de anticuerpos contra residuos citrulinados.
Existen dos posibles explicaciones para la participación de estos anticuerpos en la patogénesis de AR:
La primera es que, secundario a un aumento en la cantidad de antígenos citrulinados específicos en AR, se origina una respuesta inmune específica y por ende la producción de anticuerpos específicos. Sin embargo, la presencia de proteínas citrulinadas ha demostrado ser un fenómeno presente en cualquier tejido sinovial inflamado [25]. La segunda posibilidad, es que los pacientes con AR pueden tener una respuesta humoral anormal hacia estas proteínas y producir en forma exagerada una gran cantidad de anticuerpos.
Recientemente se demostró la presencia de anticuerpos dirigidos contra la enzima PAD IV en suero de pacientes con AR [28], tanto en población Japonesa como Caucásica. Aunque poco frecuente, su presencia es altamente específica al compararse con Lupus Eritematoso Sistémico LES y sujetos sanos. Además, se logró establecer una asociación entre los títulos elevados de estos anticuerpos con una AR más severa y también se asoció fuertemente con la presencia de anticuerpos aPCC.
La formación de complejos entre PAD IV y su substrato (péptido citrulinado), podrían actuar como portador-hapteno y ser reconocidos por células B mediante su BCR ya sea especifico para PAD IV o el sustrato citrulinado. De esta forma podrían producirse anticuerpos, ya sea contra la enzima PAD IV o contra el péptido citrulinado y presentar cualquier epítope a una célula T CD4+ específica.
Residuos Citrulinados
Bajo condiciones normales sólo pocas proteínas contienen residuos citrulinados. Estos residuos citrulinados, deberán diferenciarse de la citrulina en forma libre, cuyo metabolismo inicia en intestino delgado, en donde se sintetiza y termina en riñon, donde es degradada [29]. Esta citrulina es producida indistintamente de la citrulina en forma peptidica. La generación de citrulina en forma peptídica, es dependiente de las enzimas PAD, que como parte de modificaciones postraduccionales, convierten residuos de arginina básicos en residuos neutros de citrulina. Hasta la fecha se han identificado cinco isotipos de esta enzima. Las enzimas se encuentran distribuidos en diferentes tejidos: PAD I principalmente se expresa en epidermis y el aparato reproductor femenino; PAD II en músculo esquelético, bazo, cerebro y glándulas secretoras; PAD III en los folículos pilosos, PAD IV en neutrófilos, eosinofilos y músculo esquelético; PADVI en ovarios, embriones en etapas tempranas del desarrollo y cigotos [30].
Existe una teoría que apoya la posibilidad de un aumento en la cantidad de antígenos citrulinados, ésta sugiere que la presencia de polimorfismos y más aún, la presencia de un haplotipo particular se asoció con la presencia de AR, descrito por Suzuki y cols [31]. Está compuesto por cuatro polimorfismos de un solo nucleótido (SNPs) localizados en los exones 2, 3 y 4 del gen PADI4, localizado en el cromosoma 1, región 1p36, que codifica para la enzima peptidil arginina desaminasa IV (PAD IV). La presencia de este haplotipo de susceptibilidad, en población japonesa, se asoció con una estabilidad mayor del transcrito [31,32]; hecho, que en teoría, podría conducir a un aumento en los niveles de PADIV y por ende a la citrulinización de más epítopes. Estos epítopes citrulinados se convierten entonces en un blanco para los auto-anticuerpos específicos de la AR (anticuerpos aPCC) y al generarse un mayor número de estos complejos inmunes se estaría favoreciendo la generación de esta enfermedad autoinmune. Esta teoría en parte se comprueba posterior al hallazgo de Vossenar y cols [33], al reportar que los pacientes que eran homocigotos para el haplotipo de susceptibilidad, tuvieron títulos significativamente más altos de los anticuerpos aPCC (87% vs 67%, p <0.05), comparados con los pacientes heterocigotos y homocigotos al haplotipo de no susceptibilidad. Sin embargo, aún falta comprobar que los niveles de la enzima se encuentren elevados, y más aún, niveles elevados de antígenos citrulinados en sujetos portadores del haplotipo de susceptibilidad.
Por otro lado, es importante destacar que no sólo la presencia de células que expresan estas enzimas en tejido sinovial es condición para el desarrollo de antígenos citrulinados, dado que para la activación de dichas enzimas normalmente localizadas intracelularmente, es necesario un aumento en la concentración de Ca+2 citosólico [34].
Durante la muerte celular, la integridad de la membrana plasmática se pierde, las enzimas entonces pueden salir de la célula y activarse dado que las concentraciones de Ca+2 extracelular superan el umbral de activación de dichas enzimas (10-5 mM), de esta forma, se induce la citrulinización de proteínas extracelulares o pueden activarse en el interior de la célula al aumentar el influjo de Ca+2 y de esta manera citrulinar proteínas intracelulares [35].
Por otro lado, dada la conversión de arginina a citrulina, el grupo amino de la cadena lateral cargado positivamente es cambiado por un grupo carboxilo neutro, lo que afectaría la capacidad de los péptidos para unirse covalentemente con las moléculas de HLA.
En ratones transgénicos DRB1*0401, la citrulinación de péptidos no sólo aumentó la afinidad entre el péptido y el complejo principal de histocompatibilidad (MHC), sino que también llevó a la activación de células T CD4+ [36].
Más aún, este proceso catalítico, puede originar cambios en la estructura primaria, secundaria y terciaria de las proteínas (Figura 2). Estudios in vitro, han demostrado que un alto grado de citrulinación, puede ocasionar una desnaturalización proteica [37]
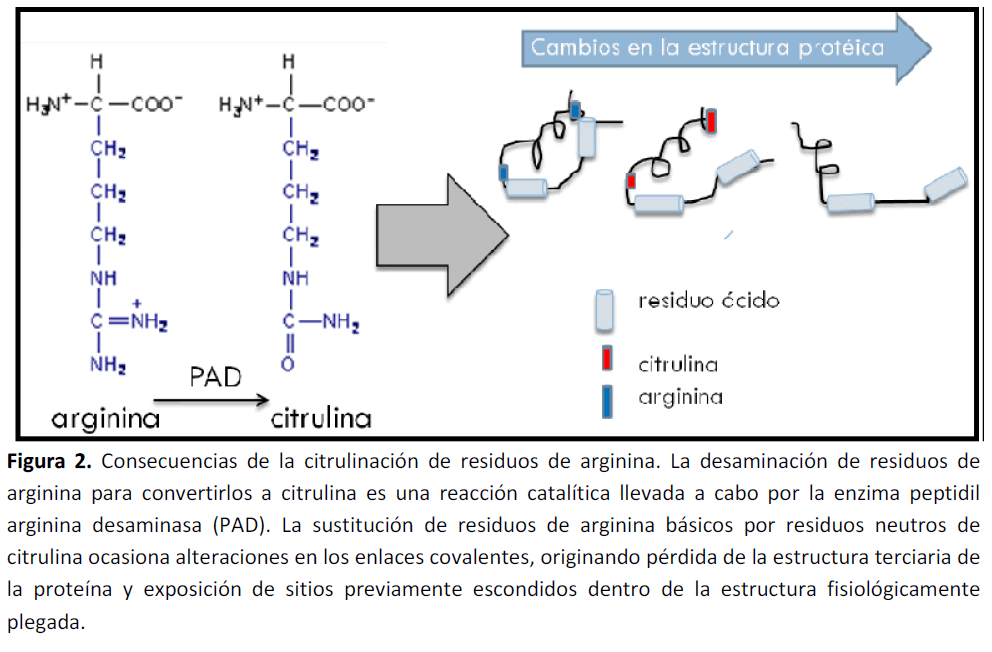
Figura 2. Consecuencias de la citrulinación de residuos de arginina. La desaminación de residuos de arginina para convertirlos a citrulina es una reacción catalítica llevada a cabo por la enzima peptidil arginina desaminasa (PAD). La sustitución de residuos de arginina básicos por residuos neutros de citrulina ocasiona alteraciones en los enlaces covalentes, originando pérdida de la estructura terciaria de la proteína y exposición de sitios previamente escondidos dentro de la estructura fisiológicamente plegada.
Pero, ¿porqué un proceso fisiológico como es la citrulinización, estaría disparando respuestas autoinmunes?
Las posibles explicaciones por las que la generación de citrulina en forma peptídica, siendo un proceso fisiológico y presente en la mayoría de los individuos es de pronto reconocido como extraño o peligroso podrían ser: 1) aumento en su producción, originando pérdida de la tolerancia por linfocitos T, 2) afectación en la capacidad de estos péptidos para unirse a moléculas del HLA con presencia del epítope compartido y 3) cambio en la estructura tridimensional de la proteína originando exposición de estructuras secundarias o primarias, antes inexpuestas al sistema inmune, o alteraciones en la unión de estas proteínas con otras.
Escape a Los Mecanismos de Tolerancia
El sistema inmune ha evolucionado para eliminar en forma eficiente lo no propio o lo peligroso según Matzinger [38]. El establecimiento y mantenimiento de la tolerancia hacia lo propio es una propiedad fundamental del sistema inmune, y la falla en esta tolerancia, conduce a la generación de autoinmunidad.
La tolerancia central implica la eliminación de células T auto-reactivas, durante su desarrollo en el timo, ya que la única clase de antígenos presentes en altas concentraciones en este órgano, son antígenos propios. De esta manera, aquellos linfocitos que reconozcan y reaccionen contra antígenos propios son eliminados (selección negativa). Sin embargo, un número de linfocitos T auto-reactivos logran escapar a esta selección y salen a la periferia. Estos linfocitos, podrían activarse y causar autoinmunidad. La tolerancia periférica, asegura que las células T auto-reactivas que escaparon los puntos de control de la tolerancia central sean eliminadas, conducidas hacia la apoptosis o sean incapaces de montar una respuesta inmune permaneciendo en estado anérgico. Los antígenos foráneos, no propios o peligrosos, son capturados y transportados hacia los órganos linfoides periféricos.
Uno de los mecanismos empleados por la tolerancia periférica es llevado a cabo mediante las células reguladoras del sistema inmune, denominadas Tregs, éstas suprimen en forma directa a las células auto-reactivas. Varias citocinas pueden influenciar la actividad de las células Tregs en forma negativa o positiva. Las promotoras principales de la actividad de las Tregs son la IL-2 y TGF-b, mientras que las citocinas que promueven la respuesta tipo Th17 (pro-inflamatoria), disminuyen la activación y funcionalidad de las Tregs [39].
La decisión del linfocito T para activarse o ser tolerante estará determinada por las características del antígeno y por la regulación en las respuestas de este linfocito T mediada por la presencia de diferentes tipos de citocinas.
El inicio y las consecuencias patológicas de las enfermedades autoinmunes, con frecuencia se ven asociadas a la presencia de alteraciones en el balance funcional entre las citocinas pro-inflamatorias (Th1 y Th17) y las inmunoreguladoras/moduladoras (Th2) [40].
Conclusiones
A pesar de los esfuerzos por reconocer el blanco de autoinmunidad en esta patología, éste aún permanece desconocido. La pregunta es, si estos residuos citrulinados presentes en exceso pudieran generar una pérdida en la tolerancia; y de ser así, ¿cuál proceso del fenómeno de tolerancia es el afectado?
Si la respuesta autoinmune se está generando hacia un proceso ocasionado secundario a muerte celular, tendríamos que preguntarnos si el reconocimiento de estos antígenos es en verdad una respuesta de tipo autoinmune o un esfuerzo del sistema inmune para deshacerse de células que se encuentran bajo situaciones de estrés.
Aún falta caracterizar la respuesta inflamatoria exacerbada presente en estos pacientes, en términos de su prioridad, es decir, ¿Es necesaria la presencia de una respuesta pro-inflamatoria para que se genere AR? o ¿La presencia de inflamación es secundaria al inicio de la enfermedad?, y lo mismo en términos de la participación de los auto-anticuerpos, ¿Son un fenómeno desencadenante? o consecuencia de la enfermedad.
El entendimiento en forma global sobre el origen, desarrollo y función de los distintos tipos celulares envueltos en la generación de autoinmunidad y su contribución para originar una pérdida de tolerancia, generarán conocimientos valiosos para el desarrollo de nuevas opciones terapéuticas.
969
References
- WJ, K., Arthritis and allied conditions. 2001, Lippincot Williams and Wilkins: Philadelphia.
- Morales-Romero, J., La atencion medica en reumatologia en un hospital de segundo nivel de atencion. Reumatol Clin, 2005. 1(2): p. 87-94.
- Sokka, T., et al., Functional disability in rheumatoid arthritis patients compared with a community population in Finland. Arthritis Rheum, 2003. 48(1): p. 59-63.
- Pearson, T.A., Where do we go from here? Am J Med, 2008. 121(10 Suppl 1): p. S32-4.
- Weyand, C.M. and J.J. Goronzy, Pathogenesis of rheumatoid arthritis. Med Clin North Am, 1997. 81(1): p. 29-55.
- Knedla, A., E. Neumann, and U. Muller-Ladner, Developments in the synovial biology field 2006. Arthritis Res Ther, 2007. 9(2): p. 209.
- Mattey, D.L., et al., Association of DRB1 shared epitope genotypes with early mortality in rheumatoid arthritis: results of eighteen years of followup from the early rheumatoid arthritis study. Arthritis Rheum, 2007. 56(5): p. 1408-16.
- van Gaalen, F.A., et al., Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum, 2004. 50(7): p. 2113-21.
- Zanelli, E., F.C. Breedveld, and R.R. de Vries, HLA class II association with rheumatoid arthritis: facts and interpretations. Hum Immunol, 2000. 61(12): p. 1254-61.
- Deighton, C.M., et al., The contribution of HLA to rheumatoid arthritis. Clin Genet, 1989. 36(3): p. 178-82.
- Takemura, S., et al., Lymphoid neogenesis in rheumatoid synovitis. J Immunol, 2001. 167(2): p. 1072-80.
- Weyand, C.M., et al., Lymphoid microstructures in rheumatoid synovitis. Curr Dir Autoimmun, 2001. 3: p. 168-87.
- Mauri, C. and M.R. Ehrenstein, Cells of the synovium in rheumatoid arthritis. B cells. Arthritis Res Ther, 2007. 9(2): p. 205.
- Firestein, G.S., Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol, 2005. 11(3 Suppl): p. S39-44.
- Nandakumar, K.S. and R. Holmdahl, Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther, 2006. 8(6): p. 223.
- van Boekel, M.A., et al., Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res, 2002. 4(2): p. 87-93.
- Zeng, X., et al., Diagnostic value of anti-cyclic citrullinated Peptide antibody in patients with rheumatoid arthritis. J Rheumatol, 2003. 30(7): p. 1451-5.
- Schellekens, G.A., et al., The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum, 2000. 43(1): p. 155-63.
- Herold, M., et al., Anti-CCP: history and its usefulness. Clin Dev Immunol, 2005. 12(2): p. 131-5.
- Vossenaar, E.R. and W.J. van Venrooij, Citrullinated proteins: sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res Ther, 2004. 6(3): p. 107-11.
- Schellekens, G.A., et al., Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest, 1998. 101(1): p. 273-81.
- Nielen, M.M., et al., Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum, 2004. 50(2): p. 380-6.
- Kastbom, A., et al., Anti-CCP antibody test predicts the disease course during 3 years in early rheumatoid arthritis (the Swedish TIRA project). Ann Rheum Dis, 2004. 63(9): p. 1085-9.
- van Gaalen, F.A., et al., Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum, 2004. 50(3): p. 709-15.
- Vossenaar, E.R., et al., The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum, 2004. 50(11): p. 3485-94.
- 26.Makrygiannakis, D., et al., Citrullination is an inflammation-dependent process. Ann Rheum Dis, 2006. 65(9): p. 1219-22.
- Verpoort, K.N., et al., Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum, 2006. 54(12): p. 3799-808.
- Takizawa, Y., et al., Peptidylarginine deiminase 4 (PADI4) identified as a conformation-dependent autoantigen in rheumatoid arthritis. Scand J Rheumatol, 2005. 34(3): p. 212-5.
- Curis, E., et al., Almost all about citrulline in mammals. Amino Acids, 2005. 29(3): p. 177-205.
- Vossenaar, E.R., et al., PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays, 2003. 25(11): p. 1106-18.
- Suzuki, A., et al., Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet, 2003. 34(4): p. 395-402.
- Harney, S.M., et al., Genetic and genomic studies of PADI4 in rheumatoid arthritis. Rheumatology (Oxford), 2005. 44(7): p. 869-72.
- Vossenaar, E.R., A.J. Zendman, and W.J. Van Venrooij, Citrullination, a possible functiona l link between susceptibility genes and rheumatoid arthritis. Arthritis Res Ther, 2004. 6(1): p. 1-5.
- Luo, Y., et al., Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry, 2006. 45(39): p. 11727-36.
- Vossenaar, E.R., et al., Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis, 2004. 63(4): p. 373-81.
- Hill, J.A., et al., Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol, 2003. 171(2): p. 538-41.
- Tarcsa, E., et al., Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J Biol Chem, 1996. 271(48): p. 30709-16.
- Matzinger, P., The danger model: a renewed sense of self. Science, 2002. 296(5566): p. 301-5.
- La Cava, A., Tregs are regulated by cytokines: implications for autoimmunity. Autoimmun Rev, 2008. 8(1): p. 83-7.
- Pelanda, R. and C.A. Piccirillo, Tolerance, immune regulation, and autoimmunity: cells and cytokines that make a difference. Curr Opin Immunol, 2008. 20(6): p. 629-31.

