Research Article - (2022) Volume 16, Issue 3
Bioequivalence study of a new fixed-dose combination tablet containing irbesartan and hydrochlorothiazide in healthy volunteers
Katya Uzunova,
Andrey Petrov,
Emil Gatchev,
Velichka Pavlova,
Elena Filipova*,
Cintia Jimenez,
Krassimir Kalinov and
Toni Vekov
Tchaikapharma High Quality Medicines Inc., Science Department, 1 G.M Dimitrov Blvd, 1172 Sofia Bulgaria, Bulgaria
Medical University of Sofia, Department of Clinical Pharmacology and Therapeutics, University Hospital “Tsaritsa Yoanna-ISUL”, 8 Byalo more str, 1527 , Bulgaria
Anapharm Bioanalytics, Encuny 22, 2nd floor, 08038 Barcelona, Spain
Medistat Ltd, Mladost 1A, block 520, ap. 54, 1729 Sofia, Bulgaria
Medical University, Dean of Faculty of Pharmacy, 1 Sv. Kliment Ohriski Str, 5800 Pleven, Bulgaria
*Correspondence:
Elena Filipova, Tchaikapharma High Quality Medicines Inc., Science Department, 1 G.M Dimitrov Blvd, 1172 Sofia Bulgaria,
Bulgaria,
Email:
Received: 30-Mar-2022, Manuscript No. Iphsj-22-12699;
Editor assigned: 01-Apr-2022, Pre QC No. Iphsj-22-12699 (PQ);
Reviewed: 12-Apr-2022, QC No. QC No. Iphsj-22-12699;
Revised: 17-Apr-2022, Manuscript No. Iphsj-22-12699(R);
Published:
25-Apr-2022, DOI: 10.36648/1791-809X.16.4.931
Abstract
Background and objective: The combination antihypertensive therapy has shown greater blood pressure lowering potential as well as better adherence to the treatment. Therefore, a new generic fixed-dose combination containing irbesartan and hydrochlorothiazide was developed and rate and extend of absorption were compared with reference formulation to prove its bioequivalence in healthy volunteers.
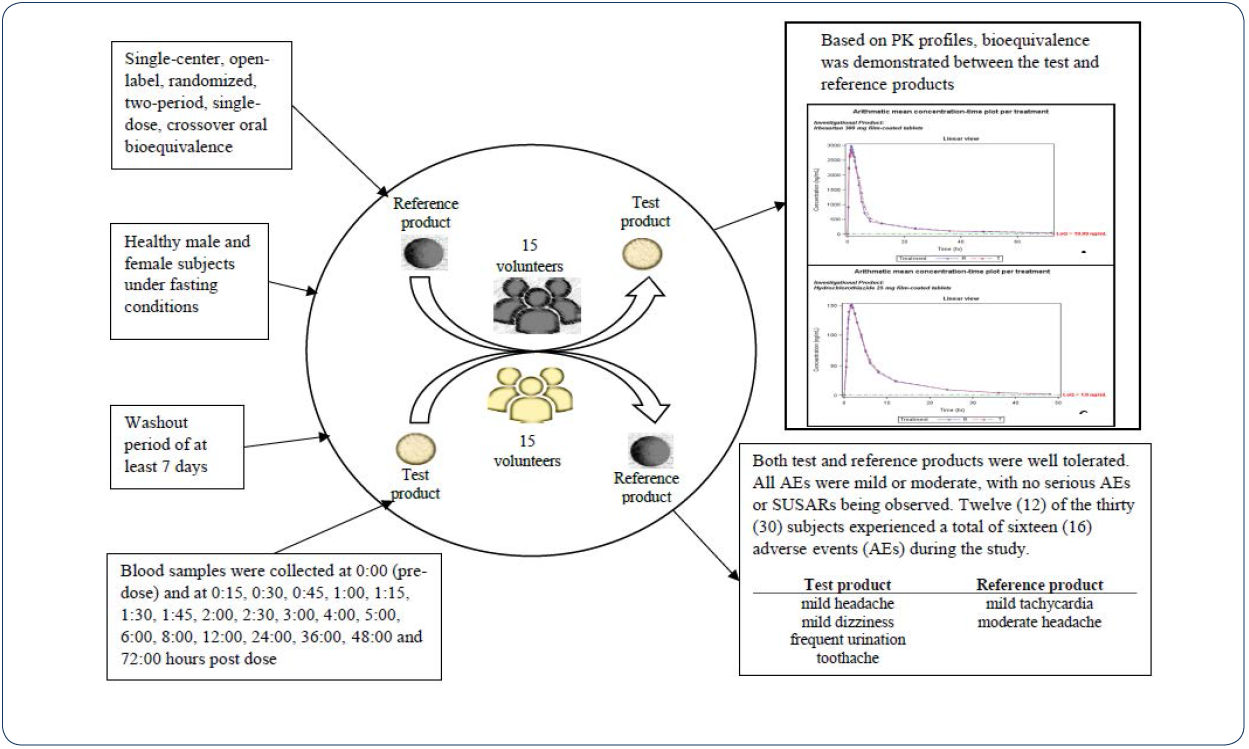
Methods: A single-centre, open-label, randomized, two-period, crossover, single dose study under fasting conditions, including at least 7-days washout period, was performed in 30 healthy male and female volunteers. Twenty blood samples were collected in each study period: prior to dosing (0:00) and up to 72:00 hours after dosing and plasma concentrations of irbesartan and hydrochlorothiazide were analysed using preliminary validated methods.
Results: The 90% CIs for the geometric mean ratios of test and reference of Cmax and AUC0–t were 89.22% to 98.80% and 100.58% to 115.11%, respectively, for irbesartan and 91.80% to 122.96% and 94.88% to 117.56%, respectively. Thus, the corresponding ratios of Cmax and AUC0-t for irbesartan and hydrochlorothiazide met the predetermined criteria for bioequivalence (90% confidence intervals of the geometric mean ratios of test and reference within the 80.00% - 125.00%). Both the test and reference products demonstrated good tolerability profile in this population, and no serious AEs were observed.
Conclusions: Therefore, the test product (Irbesartan/hydrochlorothiazide 300/25 mg film-coated tablets, manufactured by Tchaikapharma High Quality Medicines Inc., Bulgaria) and the reference product (CoAprovel 300/25 mg film-coated tablets, manufactured by Sanofi Clir SNC, France) are considered bioequivalent.
Keywords
bioequivalence; irbesartan; hydrochlorothiazide; healthy subjects
Introduction
Hypertension is a public health problem and leading cause of
mortality and disability. Pooling data from population-based
studies predicted that the global burden of hypertension would
increase by 29% by 2025, reaching close to 1.5 billion [1, 2]. Data
from the NHANES (National Health and Nutrition Examination
Survey) with 23,272 participants demonstrated higher mortality
rates among hypertensive adults than non-hypertensive adults
[3].
The high prevalence of hypertension has a substantial impact
on the burden of cardiovascular disease worldwide. In a metaanalysis
of 61 prospective observational studies each 20 mm Hg
higher systolic blood pressure (or 10 mm Hg higher diastolic blood pressure) at ages 40–69 years was associated with more than a
twofold difference in the stroke death rate, and with twofold
differences in the death rates from ischaemic heart disease (IHD)
and from other vascular causes [4]. Results from a population of
4717 hypertensive men, treated by their physicians according
to the standard clinical practice, suggest that after adjustment
for age and associated risk factors uncontrolled hypertensive
subjects presented an increased risk for cardiovascular disease
mortality and for coronary heart disease mortality compared with controlled subjects [5].
Therefore, prompt management of high blood pressure (BP)
is important to reduce the risk of target organ damage, for
prevention of cardiovascular complications, cerebrovascular
events, and death. The primary agents recommended by most
guidelines and used as monotherapy and in combination therapy
for the treatment of hypertension include thiazide diuretics,
angiotensin-converting enzyme (ACE) inhibitors, angiotensin
II receptor blockers (ARBs), and calcium channel blockers
(CCBs) [2]. Evidence from studies using fixed-dose combination
(FDC) products has shown greater BP lowering with fixed-dose
combination agents as well as better adherence to therapy [6].
The combination of ARB and thiazide diuretic results in additive
lowering of BP and was recommended for treatment of essential
hypertension [2]. A fixed-dose combination of hydrochlorothiazide
and irbesartan showed additive antihypertensive effect in a dosedependent
manner up to hydrochlorothiazide (HCTZ) 25 mg and
irbesartan 300 mg with high tolerability in diverse patient groups
[7]. BP-lowering potential and safety profile of irbesartan/HCTZ
combination were proved in clinical trials. This combination may
also be effective in indications beyond hypertension, including
congestive heart failure, post-myocardial infarction management,
diabetic nephropathy, and others [8-10].
Irbesartan has a high bioavailability (60-80%), a long duration of
action, and a small potential for pharmacological interactions due
to the nature of the enzymatic pathway involved in its metabolic
process. The peak plasma concentration after oral administration
of irbesartan occurs at 1.5-2 hours. Irbesartan is eliminated
through the liver and kidneys, with 20% of the administered dose
detectable in urine; while the rest can be found in the feces;
the elimination half-life is 11–15 hours. HCTZ is fairly rapidly
absorbed from the gastrointestinal tract. It is reported to have a
bioavailability of about 50 to 80% with peak plasma concentration
within 1 h to 2.5 h following an oral dose. Hydrochlorothiazide
is not metabolized but is eliminated rapidly by the kidneys. The
pharmacokinetics of irbesartan are not affected by the presence
of hydrochlorothiazide and vice versa [11, 12].
In the era of high prevalence of hypertension across the world,
the development of generic products that meet the criteria of
quality and effectiveness for this disease will benefit mainly
the patients. The aim the of present paper is to report the
results of a bioequivalence study of a newly developed fixeddose
generic product containing 300 mg irbesartan and 25 mg
hydrochlorothiazide compared with reference formulation in a
single-dose, 2-period, 2-sequence, crossover, and randomized
study in healthy volunteers.
Materials and Methods
The clinical part of this study was carried out at the Clinic of
Clinical Pharmacology and Therapeutics, based at the University
Hospital “TsaritsaYoanna-ISUL”, Sofia, Bulgaria. The study with
all relevant documents was reviewed and approved by the Ethics
Committee (EC) of University Hospital “TsaritsaYoanna-ISUL”,
Sofia, Bulgaria and by the Bulgarian Drug Agency. The study was
performed in accordance with the ethical principles that have
their origin in the Declaration of Helsinki (Ethical Principles for Medical Research Involving Human Subjects, last revised by the
64th WMA General Assembly, Fortaleza, Brazil, October 2013)
[13] and that are consistent with the current ICH GCP Guidelines,
regulatory requirements of the EMA [14,15] and relevant
National Laws and Regulations [16,17].
Study subjects
Prior to the screening procedure of the study, the volunteers
were informed about the nature, purpose, potential risks,
anticipated benefits, and discomforts that could arise from their
participation; and about their right to withdraw at any time.
Subjects documented their willingness to participate by signing
an informed consent form.
Eligible volunteers were healthy Caucasian subjects of both sexes
(male or female non-pregnant, non-breastfeeding, menopausal,
surgically sterile or using adequate contraception method), aged
between 18 and 55 years, BMI between 19 and 30 kg/m2, nonsmoking
or smoking up to 10 cigarettes a day, able not to smoke
from entering the clinical centre until leaving at 24th hour after
administration of the investigational product.
All eligible subjects were selected after passing screening
examination including collection of demographic data, physical
examination, laboratory tests, which included Hematology,
biochemistry, urine analysis, HIV and hepatitis B antibody and
hepatitis C antigen tests. Medical history of clinically significant
current or past diseases, surgical interventions, weight and
height measurement, body mass index calculation, vital signs
(blood pressure, heart rate, and temperature), ECG (12-lead
electrocardiogram) as well as drug abuse tests and breath tests
for alcohol were also recorded.
Investigational drug products
The following formulations were used for the study:
Irbesartan/Hydrochlorothiazide 300/25 mg film-coated tablets
manufactured by Tchaikapharma High Quality Medicines Inc.,
Bulgaria, as the test product and CoAprovel 300/25 mg filmcoated
tablets manufactured by Sanofi Clir SNC, France, as the
reference product.
Study design and drug administration
The study (EudraCT No: 2017-004862-88) was conducted as a
single-center, open-label, randomized, two-period, single-dose,
crossover oral bioequivalence study in healthy male and female
subjects under fasting conditions. Subjects (n=30) were randomly
assigned into 2 groups according to a computer-generated
randomization scheme. The volunteers were housed at the
clinical facility at least 12 hours prior to dosing in each period.
After at least a 10-hours fasting period, one tablet of the test or
the reference product was administered to each subject with 240
ml water at room temperature in each period. Following drug
administration, study subjects continued in fasting conditions
for a minimum of 4 h, and standard meals were served at
scheduled times (at 4, 8 and 12 hours post-dose for each period).
No fluid intake was allowed from 1 h before until 1 h after drug
administration. In the second period, the volunteers received the
alternate product (test or reference) after a washout period of at
least 7 days.
Blood sampling
Venous blood samples were collected into pre-labeled
vacutainers containing K2EDTA as anti-coagulating agent by a
catheter inserted into the forearm at 0:00 (pre-dose) and at 0:15,
0:30, 0:45, 1:00, 1:15, 1:30, 1:45, 2:00, 2:30, 3:00, 4:00, 5:00,
6:00, 8:00, 12:00, 24:00, 36:00, 48:00 and 72:00 hours after the
dose for the test and the reference drugs.
Following centrifugation (1900 g, 4 ± 4°C, 10 min) the separated
plasma was transferred in pre-labeled polypropylene tubes and
stored at the clinical center at -20ºC ± 5ºC until transportation to
the bio analytical laboratory (Figure 3).
Bio analytical Assay
Concentrations of irbesartan and hydrochlorothiazide in plasma
samples were analysed by Anapharm Bioanalytics, Barcelona,
Spain, using validated methods in compliance with the EMA
Guidance (EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr.
2**) [18]. Quantitative analysis for plasma concentration of
irbesartan involved a protein precipitation extraction procedure
with methanol. Irbesartan and internal standard (Irbesartan-d4)
were measured by reversed phase high performance liquid
chromatography coupled to a tandem mass spectrometry
detector (LC/MS/MS). The detection conditions of operated
mass spectrometer were positive ion mode with detection by
multiple reactive monitoring using the m/z transitions 429.20
to 207.10 for irbesartan and 433.20 to 211.10 for irbesartan-d4,
respectively. Quantitation was by peak area ratio method. A
weighted (1/X2) linear regression was performed to determine
the concentration of the analyte. The linearity of standard curve
was confirmed (r ≥ 0.9991) over the concentration range of
20.01 to 8003.20 ng/ ml with suitable accuracy and precision.
Hydrochlorothiazide is extracted from an aliquot of human EDTA
plasma using a solid-phase extraction procedure with strong
anion-exchange and reversed phase 30 mg plates and then
injected into a liquid chromatograph equipped with a tandem
mass spectrometry detector. Quantitation was by peak area
ratio method. A weighted (1/X2) linear regression was performed
to determine the concentration of the analyte. The detection
conditions of operated mass spectrometer were negative ion
mode with detection by multiple reactive monitoring using
the m/z transitions 295.80 to 268.90 for hydrochlorothiazide
and 300.70 to 270.70 for hydrochlorothiazide-15N2-13C-D2,
respectively. Calibration curves were found to be consistently
accurate and precise over the 1.01 to 302.40 ng/mL calibration
range.
Before using these methods to determine the clinical levels, the
methods were fully validated for linearity, precision, accuracy,
specificity, matrix selectivity, sensitivity, recovery, matrix effect,
dilution integrity, carryover, reinjection reproducibility, and
stability. The validated stability periods of the samples covered
the periods between the blood draws and the completion of
the analytical determination. Both studies were conducted in
compliance with the Principles of Good Laboratory Practice.
Safety assessments
The initial clinical screening was carried out not more than 30 days before the beginning of the study and included demographic
data, brief anamnestic data (medical and medication history),
physical examination, determination of body temperature,
weight and height, measurements of BP and pulse rate (PR) after
5 minutes resting in a sitting position, standard ECG (12 lead).
All of the clinical laboratory tests (albumin, alkaline phosphatase,
AST, ALT, GGT, urea, calcium, chloride, glucose, phosphorus,
potassium, serum creatinine, sodium, total bilirubin, uric
acid, total protein, cholesterol: total cholesterol, HDL, LDL,
triglycerides, as well as complete blood count with differential
count, hemoglobin, hematocrit) were performed at a contracted
and certified laboratory (medical diagnostic laboratory RAMUS).
The volunteers were also checked for the presence of HBsAg,
HCV-Ab and HIV-Ab in serum. The following parameters were
determined in urine: pH, specific gravity, protein, glucose,
ketones, bilirubin, urobilinogen, occult blood and cells, nitrite,
leukocytes. On admission, the subjects underwent tests for
alcohol, drugs, and pregnancy (urine test), while vital signs (blood
pressure, heart rate, temperature) were also measured.
All volunteers were under continuous supervision by qualified
medical staff throughout their stay in the clinical facility to
ensure their safety and wellbeing. During both study periods, the
vital signs (blood pressure and heart rate) of each volunteer were
periodically measured. Throughout the study, the physicians
questioned subjects periodically about symptoms of possible
adverse events (AEs), and subjects were encouraged to report any
unusual symptoms. All volunteers were subjected to a post-study
examination and laboratory tests on the day of the last sampling
in the second period or not more than 14 days thereafter. All
AEs, independently of their intensity, seriousness, and causality
to the investigated drugs, were recorded in source data forms
and transferred in case report forms of the volunteers.
Pharmacokinetic parameters and statistical
analysis
For the purpose of bioequivalence analysis, the primary
pharmacokinetic parameters were AUC0–t (area under the plasma
concentration-time curve from zero to the last measurable
concentration) calculated by the linear trapezoidal rule and
Cmax (maximum drug concentration in plasma) observed directly
from experimental data. Additional evaluated pharmacokinetic
parameters were Tmax (time of maximum concentration), AUC0–∞ (area under the plasma concentration-time curve from zero to
infinity), t1/2 (terminal half-life time), Kel (terminal rate constant of
elimination), residual area under the concentration-time curve.
The test product was compared to the reference product with
respect to the primary pharmacokinetic parameters using an
analysis of variance (ANOVA) with sequence, subject (sequence),
product and period effects as fixed effects after logarithmic
transformation of the data. Bioequivalence assessment was
based on a predefined acceptance criterion of 80–125% for the
90 % confidence interval for the ratio of the test and reference
products for the log-transformed data of AUC0–t and Cmax. The
statistical significance was established at p ≤ 0.05 for all statistical
tests. The data listings, descriptive statistics, statistical analysis
and graphical presentations were generated with SAS/STAT package. WinNonLin package was used for pharmacokinetic
computations (Table 3).
|
Parameter |
T/R Ratio |
90% CI (%) |
Intrasubject |
|
|
(%) |
|
(%) |
| Irbesartan |
AUC0-t |
107.6 |
100.58- 115.11 |
31.61 |
| - |
Cmax |
93.89 |
89.22- 98.80 |
25.08 |
| HCTZ |
AUC0-48 |
105.62 |
94.88- 117.56 |
12.49 |
| - |
Cmax |
106.24 |
91.80- 122.96 |
12.98 |
Table 3. Bioequivalence assessment summary for irbesartan (300 mg) and hydrochlorothiazide (25 mg).
Based on a bioequivalence range from 80.0 to 125.0% for Cmax
and AUC0-t, a within-subject coefficient of variation (CV%) not
greater than 20%, and a "test/reference" mean ratio of 93%, 28
subjects were needed to achieve a power of 80% at an alpha level
of 0.05 to show bioequivalence. To account for possible dropouts/
withdrawals additional 2 subjects were included in the
study, and as a result, thirty (30) subjects were enrolled.
Results
Study population
Тhirty (30) volunteers were included (17 males and 13 females) and were randomized into the study. One subject was withdrawn due to an observed adverse event with concomitant medication intake; hence twenty-nine (29) completed the crossover design receiving a single dose of both formulations and were included in the pharmacokinetic analysis (Figure 1). The discontinued subject was dosed only with a test product during the first period and was withdrawn before the 72 h blood sample in period 1. Demographic data of the subjects included in the study are presented in (Table 1).
| n=30 |
Age,
years |
Height,
cm |
Weight,
kg |
BMI,
kg/m2 |
| Mean |
39.10 |
169.63 |
70.80 |
24.62 |
| SD |
9.87 |
10.87 |
10.83 |
3.05 |
| %CV |
25.23 |
6.41 |
15.29 |
12.39 |
| Min |
18 |
149 |
51.8 |
19.77 |
| Max |
55 |
198 |
98 |
29.91 |
Table 1. Descriptive statistics of demographic data.
Figure 1 Disposition of the volunteers.
Safety
Twelve (12) of the thirty (30) subjects experienced a total of sixteen (16) adverse events (AEs) during the study. The reported adverse events during treatment with test product were mild headache (subjects 005 and 010) and mild dizziness (subject 001), possibly related to the treatment; as well as mildly represented frequent urination (subjects 016 and 026) considered as probably related to the treatment. Moderate to severe toothache unrelated to the administered test drug was reported by one subject twice during period 1 (subject 003).
During treatment with a reference product, were reported mild tachycardia possibly related to the administered drug and mild abdominal pain not related to the study drug. Furthermore, a moderate headache was observed (subjects 013), which was considered possibly related to the study drug and needed a concomitant treatment of paracetamol 500 mg.
Both test and reference products were generally well tolerated after single-dose administration. All AEs were mild or moderate, with no serious AEs or SUSARs (suspected unexpected serious adverse reactions) being observed. Most of the subjects who reported having an AE recovered spontaneously within a few hours or a few days of drug administration.
There was only one withdrawal during the study. Subject 003, who experienced moderate to severe toothache after check out at 24 h of Period 1, was treated with paracetamol 500 mg and lidocaine 50 mg. The adverse event was deemed unrelated to the study medications and it was resolved by tooth extraction. The subject was withdrawn by decision of the principal investigator because of possibility of pharmacokinetic profile altering interaction of the concomitant medication applied and the study drug.
Pharmacokinetic Analysis
The validated bio analytical methods were successfully applied to evaluate the bioequivalence of two tablet formulations of Irbesartan/Hydrochlorothiazide in healthy volunteers: Irbesartan/ Hydrochlorothiazide film-coated tablets of Tchaikapharma High Quality Medicines Inc., Bulgaria (test product) were compared with CoAprovel 300/25 mg film-coated tablets manufactured by Sanofi Clir SNC, France (reference product). (Figure 2)
Table 2 present the mean values and standard deviations for primary and additional pharmacokinetic parameters and geometric means for Cmax and AUC0–t. In the case of irbesartan, the observed average Cmax and AUC0–t were 3329.96±901.28 ng/mL and 22126.34±7193.63 h.ng/mL for the test product and 3511.12±945.84 ng/mL and 20299.49±7035.14 h.ng/ mL for the reference product, respectively. In the case of hydrochlorothiazide, the observed average Cmax and AUC0–t were 180.94±54.37 ng/mL and 1136.73±244.69 h.ng/mL for the test product and 174.56±60.13 ng/mL and 1092.92±297.53 h.ng/mL for the reference product, respectively. The geometric mean plasma concentration-time curves of irbesartan and hydrochlorothiazide after administration of a single dose from the test or the reference products on fasting conditions in 30 healthy volunteers are shown in figure 2 (A,C). The semi-log scale mean plasma concentration-time profiles of irbesartan and hydrochlorothiazide are shown in figure 2 (B,D) (Table 2). Bioequivalence assessment
| PK parameter |
Irbesartan |
HCTZ |
| Test |
Reference |
Test |
Reference |
| AUC0-t, ng.h/ml |
22126.34±7193.63 |
20299.49±7035.14 |
1136.73±244.69 |
1092.92±297.53 |
| |
|
|
|
|
| |
[20934.60] |
[19176.53] |
[1112.13] |
[1047.19] |
| AUC0-∞, ng.h/ml |
23652.31±8425.07 |
21549.90±7876.58 |
1167.87±249.84 |
1124.73±303.84 |
| Res,% |
5.72±4.80 |
5.18±5.18 |
2.68±1.42 |
2.95±1.41 |
| Cmax, ng/ml |
3329.96±901.28 |
3511.12±945.84 |
180.94±54.37 |
174.56±60.13 |
| |
[3214.56] |
[3389.74] |
[173.54] |
[163.00] |
| Tmax, h |
1.93±1.36 |
1.64±0.89 |
1.79±1.08 |
1.81±0.75 |
| T1/2 |
16.77±8.59 |
14.92±8.12 |
10.11±1.91 |
9.97±1.88 |
| lnCmax |
8.0800±0.2712 |
8.1300±0.2710 |
7.0140±0.2126 |
6.9539±0.3139 |
| lnAUC0-t |
9.9500±0.3483 |
9.8600±0.3444 |
5.1564±0.2934 |
5.0938±0.4006 |
Table 2. Pharmacokinetic parameters (Mean±SD) of irbesartan (300 mg) and HCTZ (25 mg) of the test and reference products [in brackets are given geometric means].
Figure 2 Arithmetic means plasma concentration–time profiles for (A) irbesartan and (C) hydrochlorothiazide; Semi-log scale
of the mean plasma concentration-time profiles for (B) irbesartan and (D) hydrochlorothiazide.
The point estimates with 90% confidence intervals of the geometric mean ratios of test and reference (T/R) in the study were found to be 93.89% (89.22 - 98.80%) for Cmax and 107.60% (100.58 - 115.11%) for AUC0-t for irbesartan and 106.24% (91.80 - 122.96%) for Cmax and 105.62% (94.88 - 117.56%) for AUC0-t for hydrochlorothiazide. Thus, the corresponding ratios of Cmax and AUC0-t met the predetermined criteria for bioequivalence (90% confidence intervals of the geometric mean ratios of test and reference within the 80.00% - 125.00%) (Table 3).
Discussion
The purpose of bioequivalence testing is to determine whether
a new formulation’s bioavailability and pharmacokinetic
parameters significantly differ from those of a reference
formulation. Therefore, the generic formulations with proven
bioequivalence and safety profile to branded products could
substitute the available reference products without repeating
clinical trials in patients and on the pharmaceutical market will
be registered quality and cost-effective generic drugs, analogs to
significantly more expensive reference products, after the expiry
of their patent protection.
The test product and the reference product had comparable
pharmacokinetic characteristics and well-tolerated profiles. The
elimination half-life for irbesartan was 16.77 hours and 14.92
hours for reference and test products, respectively, whereas
for hydrochlorothiazide it was 10.11 hours and 9.97 hours.
The washout period of 7 days was sufficient and no pre-dose
concentrations were shown. The study demonstrated that the
mean test AUC0–t values of 22126.34 (±7193.63) ng.h/ml and 1136.73 (±244.69) ng.h/ml for irbesartan and hydrochlorothiazide
respectively are comparable with the reference product and with
results of other studies [19, 20]. The administration of fixed-dose
combination irbesartan/hydrochlorothiazide in fasting condition
resulted in maximum plasma concentrations (Cmax) around 3300
ng/ml for irbesartan and 180 ng/ml for hydrochlorothiazide,
which were comparable with reference product; around 3500
ng/ml for irbesartan and 174 ng/ml for HCTZ. These findings are
consistent with other studies that assessed the pharmacokinetics
of irbesartan and HCTZ as monoproducts [21, 22] and as a
fixed-dose combination [19]. The Cmax values peaked in the
test and reference formulations at 1 h 56 min and 1 h 38min,
respectively, for irbesartan and 1 h 47 min and 1 h 49 min
for hydrochlorothiazide. All AEs were mild or moderate and
spontaneously recovered, and there were no serious AEs. Hence,
both the test and reference products were well tolerated.
In the present study, the average AUC0-t for all volunteers (except one, dosed with test formulation) was a good representative
of the extent of absorption since the average AUC0-72 obtained
was found to be greater than 80% of the average AUC0-inf. The
results suggest that 90% confidence intervals of the geometric
mean ratios of test and reference were completely within the
predefined bioequivalence criteria of 80.00% to 125.00% for the
primary pharmacokinetic parameters of AUC0-t and Cmax.
Conclusions
The present study found that the test product (Irbesartan/
hydrochlorothiazide 300/25 mg film-coated tablets,
manufactured by Tchaikapharma High Quality Medicines Inc.,
Bulgaria) and the reference product (CoAprovel 300/25 mg filmcoated
tablets, manufactured by Sanofi Clir SNC, France) were
considered bioequivalent. Both the test and reference products
demonstrated a good tolerability profile in this population, and
no serious AEs were observed.
REFERENCES
- Kearney P M, Whelton M, Reynolds K, Muntner P, Whelton P K et al. (2005) Global burden of hypertension: analysis of worldwide data, Lancet 365:217-223.
Indexed at, Google Scholar, Crossref
- B Williams, G Mancia, W Spiering, E Agabiti Rosei, M Azizi et al. (2018) ESC Scientific Document Group, 2018 ESC/ESH Guidelines for the management of arterial hypertension, Eur Heart J 39:3021-3104.
Indexed at, Google Scholar, Crossref
- E S Ford (2011) Trends in mortality from all causes and cardiovascular disease among hypertensive and non-hypertensive adults in the United States Circulation 123:1737-1744.
Indexed at, Google Scholar, Crossref
- S Lewington, R Clarke, N Qizilbash, R Peto, R Collins R et al. (2002) Prospective studies collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies, Lancet 360:1903-1913.
Indexed at, Google Scholar, Crossref
- A Benetos, F Thomas, K Bean, S Gautier, H Smulyan (2002) Prognostic value of systolic and diastolic blood pressure in treated hypertensive men. Arch Intern Med 162:577-581.
Indexed at, Google Scholar, Cross Ref.
- P K Whelton, RM Carey, WS Aronow, DE Casey Jr, K J Collins et al. (2017) ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines, Circulation 138:e426-e483.
Indexed at, Google Scholar, Crossref
- G Derosa, I Ferrari, A F Cicero (2009) besartan and hydrochlorothiazide association in the treatment of hypertension. Curr Vasc Pharmacol 7:120-136.
Indexed at, Google Scholar, Cross Ref.
- J Rosenstock, L Rossi, C S Lin, D MacNeil, M Osbakken (1998) the effects of irbesartan added to HCTZ for the treatment of hypertension in patients non-responsive to HCTZ alone. J Clin Pharm Ther 23:433-440.
Indexed at, Google Scholar, Crossref
- P Raskin, R Guthrie, J M Flack, R Reeves, R Saini (1999) the long-term antihypertensive activity and tolerability of irbesartan with HCTZ. J Hum Hypertens 13:683-7.
Indexed at, Google Scholar, Crossref
- M Kochar, R Guthrie, J Triscari, K Kassler-Taub, R A Reeves (1999) Matrix study of irbesartan with HCTZ in mild-to-moderate hypertension. Am J Hypertens 12:797-805.
Indexed at, Google Scholar, Crossref
- SmPC, Co-Aprovel (2021) European medical agency-European union
Google Scholar
- C Borghi, A F Cicero (2012) the role of irbesartan in the treatment of patients with hypertension: a comprehensive and practical review. High Blood Press Cardiovasc Prev 19:19-31.
Indexed at, Google Scholar, Crossref
- WMA (1964) Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, and amended by the 64th WMA General Assembly. Fortaleza, Brazil.
Indexed at, Crossref
- EMA/CHMP/ICH/135/1995 ICH Topic E6 (R2). Guideline for Good Clinical Practice Step 5.
Google Scholar
- CPMP/EWP/QWP/1401/98 (2010) Guideline on the Investigation of Bioequivalence.
Google Scholar
- Bulgarian law on the medicinal products in human medicine: Medicinal Products in Human Medicine Act.
Google Scholar
- Ordinance No. 31 dated 17 August 2007 on the rules of good clinical practice (issued by the Ministry of Health.
Google Scholar, Crossref
- EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2 Guideline on bio analytical method validation, 21 July 2011.
Indexed at, Google Scholar, Crossref
- NL/H/2910/001-003/DC Public Assessment Report. Scientific discussion. Converide 150 mg/12.5 mg, 300 mg/12.5 mg and 300 mg/25 mg film-coated tablets, 2014.
Indexed at, Google Scholar
- M Cánovas, F Cabré, F Polonio (2014) Bioequivalence studies for 2 different strengths of irbesartan/hydrochlorothiazide combination in healthy volunteers: 300/25 mg and 300/12.5 mg film-coated tablets. Drug Res 62:257-262.
Indexed at, Google Scholar, Crossref
- NL/H/3816/001-002/DC (2017) Assessment Report. Scientific discussion. Hydrochlorothiazide Farmaprojects 12.5 mg and 25 mg tablets.
Google scholar
- EMEA/H/C/000962 (2008) CHMP ASSESSMENT REPORT FOR Irbesartan Krka.
Indexed at, Google Scholar
Citation: Uzunova K, Petrov A, Gatchev E, Pavlova V, Filipova E, et al. (2022) Bioequivalence study of a new fixed-dose combination tablet containing irbesartan and hydrochlorothiazide in healthy volunteers. Health Sci J. Vol. 16 No. 3: 931.