Atul Arya1*, Surbhi Rana1, Sumeet Gupta2 and Lakhwinder Singh3
1Department of Pharmaceutical Sciences, I. K. Gujral Punjab Technical University, Jalandhar, Punjab-144603, India
2Department of Pharmacology, Maharishi Markandeshwar University, Mullana, Ambala, Haryana-133207, India
3Department of Applied Science CGC College of Engineering, Landran, Mohali, Punjab-140307, India
- *Corresponding Author:
- Arya A
Cardiovascular Division, Department of Pharmaceutical Sciences
I.K. Gujral Punjab Technical University,
Jalandhar, Punjab-144603, India
Tel: +91 9779981111
E-mail: atularya07@gmail.com
Rec date: January 6, 2016, Acc date: February 24, 2016, Pub date: February 29, 2016
Copyright: © 2016 Arya A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
Endothelial nitric oxide synthase; Vascular endothelial dysfunction; Nitric oxide
Introduction
An epidemic of diabetic nephropathy
Diabetic nephropathy (DN) is a leading cause of mortality and morbidity in patients with diabetes. It is the single major cause of renal failure in many countries. This complication reflects a complex pathophysiology, whereby various genetic and environmental factors determine susceptibility and progression to end-stage renal disease. End-stage renal disease (ESRD) due to diabetes has been estimated to be 30-47% of all incident cases worldwide [1]. Disparities in the incidence of ESRD from diabetes among ethnic groups have existed for many years, but the magnitude has been increasing. The World Health Organization (WHO) has estimated that there are currently 346 million people affected by diabetes worldwide and anticipates that diabetes-related deaths would double by 2030 [2]. These figures highlight the importance of continued research and the need for novel methods to both prevent and treat this pandemic [3].
There is compelling evidence that endothelial dysfunction serves as a key event in the development and progression of diabetic vascular complications, including nephropathy [4-6]. Endothelial cells maintain vascular function and homeostasis by generating paracrine factors that regulate vascular tone, preventing coagulation and platelet aggregation, inhibiting adhesion of leukocytes, and limiting proliferation of vascular smooth muscle cells as well as by constituting a selective barrier to the diffusion of macromolecules into the interstitial space. Further, it was shown that Nitric Oxide (NO) produced by endothelial cells through the endothelial Nitric Oxide Synthase (eNOS) plays a major role for many of these endothelial functions [5] and that decreased NO production and bioavailability largely contribute to endothelial dysfunction in diabetes [6].
The endothelium
Endothelium is a type of epithelium that lines the interior surface of blood vessels and lymphatic vessels. Vascular endothelium is an innermost layer of blood vessels responsible for regulation of vascular tone and free flow of blood in vessels [7].
The multiple functions of vascular endothelium are summarized in Figure 1 and include regulation of vessel integrity, vascular growth and remodeling, tissue growth and metabolism, immune responses, cell adhesion, angiogenesis, hemostasis and vascular permeability. The endothelium plays a pivotal role in the regulation of vascular tone, controlling tissue blood flow and inflammatory responses and maintaining blood fluidity [8,9].
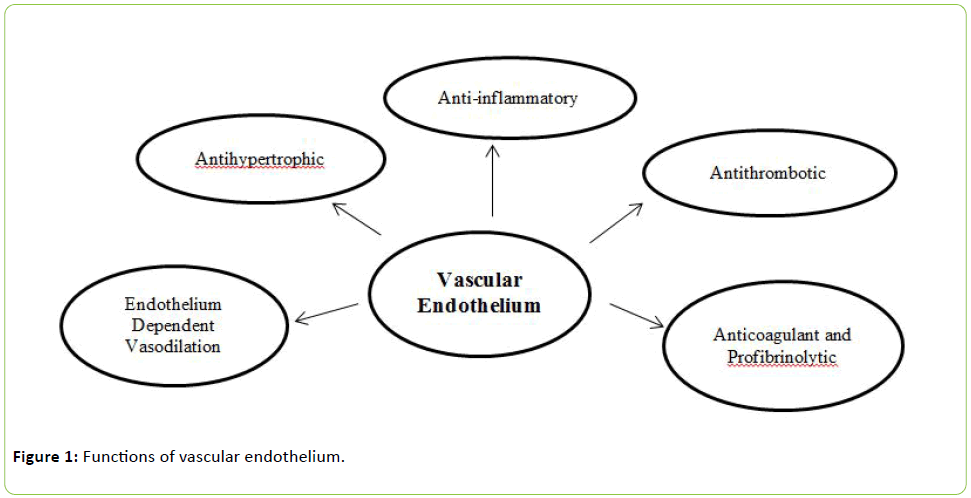
Figure 1: Functions of vascular endothelium.
Vascular endothelium is the inner layer of the blood vessels, which serves as an important role to regulate the vascular functions and blood flow in vessels [7].
Vascular endothelium maintains the vascular tone and exerts anticoagulant, antithrombotic, antiplatelet and fibrinolytic properties [10]. Endothelium is a multifunctional organ and it control the release of endothelium derived relaxing factors (EDRF), endothelium derived contracting factors (EDCF), endothelium derived hyperpolarizing factors (EDHF) [11,12] inflammatory mediators such as intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), nuclear factor kappa-B (NF-kB) and various growth factors like vascular endothelial growth factors (VEGF) and transforming growth factor-β (TGF-β) [4,11].
Nitric oxide
Nitric oxide is a small gaseous and lipophilic molecule that participates of several biological processes. It is synthesized from Endothelial NOS (eNOS), also known as nitric oxide synthase 3 (NOS3) or constitutive NOS (cNOS), is an enzyme that in humans is encoded by the NOS3 gene located in the 7q35-7q36 region of chromosome 7 [13]. Nitric oxide (NO) is an important protective molecule in the vasculature, and endothelial NO synthase (eNOS) is responsible for most of the vascular NO produced.
eNOS has a protective function in the cardiovascular and renal system, which is attributed to NO production.
Regulation of the vascular tone: Once the nitric oxide diffuses from the endothelial cells, it activates the enzyme soluble guanylate cyclase (sGC), which catalyses the conversion of guanosine triphosphate into cyclic guanosine monophosphate (cGMP) which in turn activates protein kinase G (PKG) and promotes the phosphorylation of cellular targets and promotes the vascular relaxation.
Antiproliferative property: NO exerts antiproliferative effects by cGMP-dependent inhibiting Ca2+ influx or by directly inhibiting the activity of arginase and ornithine decarboxylase, decreasing the generation of polyamides required for DNA synthesis [14,15].
Antithrombotic effects: Nitric oxide exerts antithromic effects by diffusing across the platelet membrane and resulting in inhibition of platelet aggregation.
Leukocyte adhesion: Nitric oxide (NO) is a biologically active compound produced by vascular endothelium and is rapidly inactivated by superoxide. There is circumstantial evidence in the literature that NO may interfere with the ability of polymorphonuclear leukocytes to adhere to microvascular endothelium [16]. It is well established that NO prevents the adhesion of platelets to endothelial monolayers. Additionally, NO inhibits neutrophil aggregation in vitro, an effect that is potentiated by superoxide dismutase. Moreover, NO affects leukocyte adhesion to the vascular endothelium by inhibiting the nuclear factor kappa B (NF-κB), which induces vascular endothelial expression of chemokines and adhesion molecules [17,18].
Attributable to up-regulation of heme-oxygenase-I and ferritin expression, which reduce superoxide anion concentrations in blood vessels [19].
Vascular endothelial dysfunction
In patients with diabetes, endothelial dysfunction appears to be a consistent finding; indeed, there is general agreement that hyperglycemia and diabetes lead to an impairment of NO production and activity. Endothelial dysfunction is a systemic pathological condition which can be broadly defined as an imbalance between vasodilating and vasoconstricting substances produced by the endothelium or overall functions of the endothelium [20]. Normal functions of endothelial cells include production of nitric oxide (NO), regulation of platelet adhesion, coagulation, immune function, control of volume, and electrolyte content of the intravascular and extravascular spaces [21,22]. Endothelial dysfunction is primarily due to reduction in NO bioavailability, and a marker for vascular health. Endothelial dysfunction can result from and/or contribute to several disease processes, as occurs in diabetes mellitus, hypercholesterolemia and hypertension, and also due to environmental factors, such as smoking tobacco products and exposure to air pollution [23-25].
Additionally, endothelial dysfunction is characterized by one or more of the following features: reduced endotheliummediated vasorelaxation, hemodynamic deregulation, impaired fibrinolytic ability, enhanced turnover, overproduction of growth factors, increased expression of adhesion molecules and inflammatory genes, excessive generation of ROS, increased oxidative stress, and enhanced permeability of the cell layer [26,27].
Vascular endothelial dysfunction cause reduced activation of endothelial nitric oxide synthase, decreased generation and bioavailability of nitric oxide (NO). VED leads to increase in production of reactive oxygen species (ROS) and proinflammatory mediators are implicated in pathogenesis of various disorders like hypertension, atherosclerosis and diabetic nephropathy [28-30].
Diabetic nephropathy and vascular endothelial dysfunction
Hyperglycemia causes vascular endothelial dysfunction via micro and macrovascular complications [31]. Diabetic nephropathy is defined as partial loss of nephrons followed by nephrotic syndrome and glomerulosclerosis. Nephropathy is characterized by persistent elevated albuminuria, declined glomerular filtration rate, and elevated arterial blood pressure and oedema [32,33]. Although several other factors may mediate in the development and progression of diabetic nephropathy.
Insulin resistance usually precedes the development of Type II diabetes and is often accompanied by a cluster of cardiovascular risk factors, notably obesity, hypertension, high triacylglycerol (triglyceride) levels, low HDL (high-density lipoprotein)-cholesterol levels, abnormal LDL composition, hyperinsulinaemia, insulin resistance and inflammation, all of these impair endothelial function. Hyperglycemia is the major causal factor in the development of endothelial dysfunction in diabetes mellitus [30].
Diabetes mellitus (DM) is a complex metabolic syndrome characterized by absolute insulin deficiency or development of insulin resistance that leads to altered glucose, fat and protein metabolism [34]. Retinopathy, neuropathy, cardiomyopathy, and nephropathy are long term complications due to diabetes mellitus and endothelial nitric oxide synthase imbalance. Both genetic and environmental factors are involved in the development of endothelial dysfunction during DN [35].
High concentration of glucose has been noted to scavenge nitric oxide and induce vascular endothelial dysfunction finally leads to diabetic nephropathy [36] which is followed by formation of advance glycation end-products (AGEs), reactive oxygen species (ROS) and increase in the oxidative stress [37,38].
Nevertheless, there is no doubt that chronic hyperglycemia and the subsequent metabolic derangements play a major role in diabetic EC injury and can lead to the over production of advanced glycation end products (AGE), activated protein kinase C (PKC) signaling cascades and accumulated reactive oxygen species (ROS) [39]. Hemodynamic alterations along with renin-angiotensin system (RAS) regulation seem to be another pivotal contributor to dysfunction of renal endothelium in both glomerular afferent and efferent arteries [40].
Diabetic nephropathy which is characterized by glomerular hypertrophy, accumulation of extracellular matrix protein, increased basement membrane thickness, mesangial expansion, podocyte loss, and vascular endothelial dysfunction progressively leading to glomerulosclerosis, tubulointerstitial fibrosis, and proteinuria [41].
It has been noted in various studies that hyperglycemia induce endothelium cell death by activating bax-caspase proteases pathway [42].
Further, diabetic nephropathy is associated with activation of various intracellular signaling mechanisms and transcription factors, i.e. protein kinase C (PKC), mitogen activated protein kinase (MAPKs), nuclear factor kappa B (NFκB) [43]. Furthermore, overexpression of various growth factors, i.e. transforming growth factor (TGF-β), vascular endothelial growth factor (VEGF), and cytokines, i.e. tumor necrosis factor a (TNF-α), interleukin 1 (IL1), and insulin-like growth factor-1 (IGF-1), stimulates proliferation of mesangial cells contributing to glomerulosclerosis and tubulointerstitial fibrosis [44].
Various endogenous modulators such as Ang-II, ET-I, caveolin, resistin and Rho-kinase are upregulated in VED [45,46] whereas adiponectin and apelin are downregulated in VED [47]. Ang-II stimulates the release of VEGF and develops proteinuria in diabetic nephropathy [45].
The experimental evidences suggests that hyperlipidemia may mediate renal injury by increasing the expression of sterol regulatory element-binding protein (SREBP), which is responsible for increasing the synthesis of triglycerides and cholesterol in the kidney, that are associated with increased expression of TGF-β, VEGF, extracellular matrix proteins, type IV collagen and fibronectin resulting in glomerular hypertrophy. Further, SREBP stimulates podocyte injury, glomerulosclerosis and tubulointerstitial fibrosis to produce nephropathy [48,49].
In addition, diabetes upregulates the generation of AGEs, which contributes to endothelial cell death and vascular endothelial dysfunction [50]. Moreover, the expression and activity of eNOS is down regulated through glucose mediated production of reactive oxygen species (ROS) in diabetes [51]. The ROS thus generated during diabetes play a major role in the pathogenesis of diabetic nephropathy [52]
Oxidative stress and endothelial cell dysfunction
Oxidative stress describes the condition wherein an excessive production of ROS overwhelms endogenous antioxidant defense mechanisms. Oxidative stress is caused by three factors: (1) an increase in oxidant generation, (2) a decrease in antioxidant protection, (3) a failure to repair oxidative damage [53-55]. There are numerous risk factors that can cause endothelial cell damage under diabetes such as hyperglycemia, insulin resistance, dyslipidemia, increased oxidative stress, inflammation, and hypertension [56,57].
The vascular endothelium, which regulates the passage of macromolecules and circulating cells from blood to tissues, is a major target of oxidative stress, playing a critical role in the pathophysiology of several vascular diseases and disorders [58]. Specifically, oxidative stress increases vascular endothelial permeability and promotes leukocyte adhesion, which is coupled with alterations in endothelial signal transduction and redox-regulated transcription factors [59].
Endothelial dysfunction is associated with decreased NO availability, either through loss of NO production or through loss of NO biological activity [60]. NO production is diminished in cells which are subject to oxidative stress. Hyperglycemiainduced eNOS impairment leads to increased oxidative stress and scavenging of NO, which represents initiation event(s) for development of endothelial dysfunction [61].
A decline in NO bioavailability may be caused by decreased expression of the endothelial cell NO synthase (eNOS), a lack of substrate or cofactors for eNOS, alterations of cellular signaling such that eNOS is not appropriately activated and finally accelerate NO degradation by ROS [62-65].
The imbalance between NO and reactive oxygen species (ROS) generation is a central pathophysiologic denominator in diabetic endothelial dysfunction. High glucose increases ROS production in ECs [66] and reduces endogenous antioxidant systems [67] resulting in oxidative stress.
It is known that under diabetic conditions there are increased oxidative stress levels [68]. Increased ROS prompts the EPCs to produce pathologic cytokines such as monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor-α (TNF-α), NF-κB, interleukin-8 (IL-8), elevated levels of iNOS, and decreased eNOS. Diabetes which is a metabolic disorder characterized by impaired endogenous insulin secretion and activity, reduced NO production and increased production of free radicals, or impaired antioxidant defenses. The predominant factor in diabetes-mediated complications is endothelial dysfunction. The mechanisms that lead to endothelial dysfunction in diabetes are complex [69,70].
Both endothelial cells and vascular smooth muscle cells are capable of producing reactive oxygen species from a variety of enzymatic sources. In disease states such as diabetes, vascular production of reactive oxygen metabolites can increase substantially [68]. Increased production of the superoxide anion can lead to decreased tissue bioavailability of nitric oxide (NO) via a facile radical/radical reaction that occurs more rapidly than the reaction of with superoxide dismutase (SOD) [71]. This phenomenon alters endothelial regulation in a variety of disease conditions. Importantly, this endothelial dysfunction is due to vascular production of superoxide. There are several enzymes that involve generating ROS such as NADPH oxidase, aldehyde oxidase, xanthine oxidase, and glucose oxidase [68]. Decreased endothelium-dependent vasodilation in diabetic subjects is associated with the impaired action of NO secondary to its inactivation resulting from increased oxidative stress, rather than decreased NO production from vascular endothelium and that abnormal NO metabolism is related to advanced diabetic microvascular complications [72].
Potential Therapeutic Strategies
The risk of cardiovascular death in diabetic patients with microalbuminuria is some 7-40 times that of an age matched general population. Lifestyle changes are required to prevent the onset of diabetes. They include cessation of smoking and require regular exercise, weight loss and controlled sodium intake.
Glycemic control and Renin Angiotensin Aldosterone system (RAAS) inhibition have long been mainstays of therapy in patients with diabetic nephropathy [73].
The natriuretic peptides (NPs), which are secreted from cardiomyocytes in response to cardiac wall stress, play an important role in the regulation of blood pressure, intravascular volume, and cardiac remodeling. The NPs consists of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C type natriuretic peptide, where ANP and BNP are secreted by the heart in response to increased volume and pressure load [74].
Natriuretic peptides are mainly involved in water and sodium balance and cardiovascular homeostasis, several neurohormones such as endothelin-1 (ET-1), Catecholamines stimulate the secretion of natriuretic peptides [75,76]. The effects of ANP are mediated by the transmembrane guanylylcyclase receptor type A, which promotes intracellular cGMP formation [77].
Administration of ANP drugs (5 μg/kg/i.p.) and (10 μg/kg/ i.p.) inhibit the renin-angiotensin II-aldosterone system [78].
Diabetic nephropathy is a potentially fatal endpoint of uncontrolled diabetes. Reduced level of nitric oxide is most negative factor involved in diabetic nephropathy. In hyperglycemia due to up regulation of endothelial dysfunction mediators such as Ang-II, Endothelin-I, Caveolin are actively involved in diabetic nephropathy [79].
Regarding the endothelial dysfunction, Single therapy may not adequately improve endothelial function, so it is necessary to target multiple factors for therapeutic intervention of endothelial dysfunction. Targeting more than one risk factor of endothelial damage only can improve endothelial functions.
Treatments that improve endothelial function systemically, like ACE inhibitors, statins, metformin, antioxidants, folate, PKC-inhibitors, and supplements like L-arginine, BH4, folic acid, and polyphenols also appear to provide protection from diabetes mediated vascular events [80].
10747
References
- Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, et al. (2013) Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol 24: 302-308.
- World Health Organization and C. D. Bode (2011) Media Centre-Fact Sheets, Diabetes.
- Fowler MJ (2008) Microvascular and macrovascular complications of diabetes. Clinical Diabetes 26: 77-82.
- Xu J, Zou MH (2009) Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 120: 1266-1286.
- Rask-Madsen C, King GL (2007) Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab 3: 46-56.
- Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S (2009) Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 32 Suppl 2: S314-321.
- Vallance P (2001) Importance of asymmetrical dimethylarginine in cardiovascular risk. Lancet 358: 2096-2097.
- Bonetti PO, Lerman LO, Lerman A (2003) Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol 23: 168-175.
- Félétou M, Vanhoutte PM (2006) Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 291: H985-1002.
- Stehouwer CD (2004) Endothelial dysfunction in diabetic nephropathy: state of the art and potential significance for non-diabetic renal disease. Nephrol Dial Transplant 19: 778-781.
- Garland CJ, Hiley CR, Dora KA (2011) EDHF: spreading the influence of the endothelium. Br J Pharmacol 164: 839-852.
- Calles-Escandon J, Cipolla M (2001) Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev 22: 36-52.
- Marsden PA, Schappert KT, Chen HS, Flowers M, Sundell CL, et al. (1992) Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett 307: 287-293.
- Cornwell TL, Arnold E, Boerth NJ, Lincoln TM (1994) Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am J Physiol 267: C1405-1413.
- Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, et al. (2001) Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A 98: 4202-4208.
- Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, et al. (2000) Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631-1639.
- Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, et al. (1992) Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267: 18148-18153.
- Walford G, Loscalzo J (2003) Nitric oxide in vascular biology. J Thromb Haemost 1: 2112-2118.
- Chen F, Castranova V, Shi X, Demers LM (1999) New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem 45: 7-17.
- Goligorsky MS (2005) Endothelial cell dysfunction: can't live with it, how to live without it. Am J Physiol Renal Physiol 288: F871-880.
- Deanfield J, Donald A, Ferri C (2005) Endothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the working group on endothelin and endothelial factors of the European society of hypertension. Journal of Hypertension 23: 7-17.
- Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27: 1047-1053.
- Li H, Isomaa B, Taskinen MR, Groop L, Tuomi T (2000) Consequences of a family history of type 1 and type 2 diabetes on the phenotype of patients with type 2 diabetes. Diabetes Care 23: 589-594.
- Steinberger J, Daniels SR (2003) Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American heart association scientific statement from the atherosclerosis, hypertension, and obesity in the young committee (council on cardiovascular disease in the young) and the diabetes committee (council on nutrition, physical activity, and metabolism) Circulation 107: 1448-1453.
- Bir SC, Esaki J, Marui A, Yamahara K, Tsubota H, et al. (2009) Angiogenic properties of sustained release platelet-rich plasma: characterization in-vitro and in the ischemic hind limb of the mouse. J Vasc Surg 50: 870-879.
- Taddei S, Ghiadoni L, Virdis A, Versari D, Salvetti A (2003) Mechanisms of endothelial dysfunction: clinical significance and preventive non-pharmacological therapeutic strategies. Curr Pharm Des 9: 2385-2402.
- Laughlin MH, Newcomer SC, Bender SB (2008) Importance of hemodynamic forces as signals for exercise-induced changes in endothelial cell phenotype. J Appl Physiol (1985) 104: 588-600.
- Cade WT (2008) Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys Ther 88: 1322-1335.
- Addabbo F, Montagnani M, Goligorsky MS (2009) Mitochondria and reactive oxygen species. Hypertension 53: 885-892.
- Hirose A, Tanikawa T, Mori H, Okada Y, Tanaka Y (2010) Advanced glycation endproducts increase endothelial permeability through the RAGE/Rho signaling pathway. FEBS Lett 584: 61-66.
- Michael J, Fowler (2008) Microvascular and Macrovascular Complications of Diabetes. Clinical Diabetes 26: 77-82.
- Takizawa T, Takasaki I, Shionoiri H, Ishii M (1997) Progression of glomerulosclerosis, renal hypertrophy and an increased expression of fibronectin in the renal cortex associated with aging and salt-induced hypertension in Dahl salt-sensitive rats. Life Sci. 61: 1553-1558.
- Blicklé JF, Doucet J, Krummel T, Hannedouche T (2007) Diabetic nephropathy in the elderly. Diabetes Metab 33 Suppl 1: S40-55.
- Ichinose K, Kawasaki E, Eguchi K (2007) Recent advancement of understanding pathogenesis of type 1 diabetes and potential relevance to diabetic nephropathy. Am J Nephrol 27: 554-564.
- Kolluru GK, Bir SC, Kevil CG (2012) Endothelial Dysfunction and Diabetes: Effects on Angiogenesis, Vascular Remodeling, and Wound Healing. International Journal of Vascular Medicine 30.
- Kanwar YS, Sun L, Xie P, Liu FY, Chen S (2011) A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol 6: 395-423.
- Futrakul N, Panichakul T, Sirisinha S, Futrakul P, Siriviriyakul P (2004) Glomerular endothelial dysfunction in chronic kidney disease. Ren Fail 26: 259-264.
- Zhao HJ, Wang S, Cheng H, Zhang, Z, Takahashi T, et al. (2006) Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664-2669.
- Ansar S, Koska J, Reaven PD (2011) Postprandial hyperlipidemia, endothelial dysfunction and cardiovascular risk: focus on incretins. Cardiovasc Diabetol 10: 61.
- Arcaro G, Cretti A, Balzano S, Lechi A, Muggeo M, et al. (2002) Insulin causes endothelial dysfunction in humans: sites and mechanisms. Circulation 105: 576-582.
- Nakagami H, Kaneda Y, Ogihara T, Morishita R (2005) Endothelial dysfunction in hyperglycemia as a trigger of atherosclerosis. Curr Diabetes Rev 1: 59-63.
- Nakagami H, Morishita R, Yamamoto K, Yoshimura S, Taniyama Y, et al. (2001) Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes 50: 1472-1481.
- Tomlinson DR (1999) Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia 42: 1271-1281.
- Navarro-González JF, Mora-Fernández C (2008) The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433-442.
- d’Uscio LV, Shaw S, Barton M, Luscher TF (2006) Losartan but not verapamil inhibits angiotensin-II-induced tissue endothelin-I increase role of blood pressure and endothelial function. Hypertension 31: 1305-1310.
- Lee EY, Shim MS, Kim MJ, Hong SY, Shin Y, et al. (2004) Angiotensin II receptor blocker attenuates overexpression vascular endothelial growth factor in diabetic podocytes. Exp Mol Med 36: 65-70.
- Ouchi N, Walsh K (2007) Adiponectin as an anti-inflammatory factor. Clin Chim Acta 380: 24-30.
- Wang Z, Jiang T, Li J, Proctor G, McManaman JL, et al. (2005) Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes 54: 2328-2335.
- Wolf G, Ziyadeh FN (2007) Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol 106: p26-31.
- Shrinivasan S, Hatley ME, Bolick DT, Palmer LA, Edelstein D, et al. (2004) Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 47: 1727-1734.
- Satchell SC, Tooke JE (2008) What is the mechanism of microalbuminuria in diabetes: a role for the glomerular endothelium? Diabetologia 51: 714-725.
- Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, et al. (1998) Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A 95: 9220-9225.
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, et al. (2010) S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115-1118.
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239-247.
- Dandona P, Aljada A (2004) Endothelial dysfunction in patients with type 2 diabetes and the effects of thiazolidinedione antidiabetic agents. Journal of Diabetes and Its Complications 18: 91-102.
- Hadi HA, Suwaidi JA (2007) Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 3: 853-876.
- Lum H, Roebuck KA (2001) Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol 280: C719-741.
- Hedner T, Sun X (1997) Measures of endothelial function as an endpoint in hypertension? Blood Press Suppl 2: 58-66.
- Harrison DG (1997) Cellular and molecular mechanisms of endothelial cell dysfunction. Journal of Clinical Investigation 100: 2153-2157.
- Goligorsky MS, Chen J, Brodsky S (2001) Workshop: endothelial cell dysfunction leading to diabetic nephropathy : focus on nitric oxide. Hypertension 37: 744-748.
- Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, et al. (1997) Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc. Biol 17: 2479-2488.
- Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM (1992) Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem 267: 24173-24176.
- Shimokawa H, Flavahan NA, Vanhoutte PM (1991) Loss of endothelial pertussis toxin-sensitive G protein function in atherosclerotic porcine coronary arteries. Circulation 83: 652-660.
- Harrison DG (1997) Endothelial function and oxidant stress. Clin Cardiol 20: II-11-7.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemic damage. Nature 404: 787-790.
- Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, et al. (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem 279: 30369-30374.
- Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA (2010) Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm 2010: 453892.
- Witting PK, Rayner BS, Wu BJ, Ellis NA, Stocker R (2007) Hydrogen peroxide promotes endothelial dysfunction by stimulating multiple sources of superoxide anion radical production and decreasing nitric oxide bioavailability. Cellular Physiology and Biochemistry 20: 255-268.
- McCance DR, Hanson RL, Pettitt DJ, Bennett PH, Hadden DR, et al. (1997) Diagnosing diabetes mellitus--do we need new criteria? Diabetologia 40: 247-255.
- Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315-424.
- Maejima K, Nakano S, Himeno M (2001) Increased basal levels of plasma nitric oxide in Type 2 diabetic subjects. Relationship to microvascular complications. J. Diabetes Complications 15: 135-143.
- ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, Neal B, et al. (2008) Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 358: 2560-2572.
- Levin ER, Gardner DG, Samson WK (1998) Natriuretic peptides. N Engl J Med 339: 321-328.
- Irons CE, Murray SF, Glembotski CC (1993) Identification of the receptor subtype responsible for endothelin-mediated protein kinase C activation and atrial natriuretic factor secretion from atrial myocytes. J. Biol. Chem 268: 23417-23421.
- Jin HK, Chen YF, Yang RH, McKenna TM, Jackson RM, et al. (1989) Vasopressin lowers pulmonary artery pressure in hypoxic rats by releasing atrial natriuretic peptide. Am J Med Sci 298: 227-236.
- Hunt PJ, Espiner EA, Richards AM, Yandle TG, Frampton C, et al. (1995) Interactions of atrial and brain natriuretic peptides at pathophysiological levels in normal men. Am J Physiol 269: R1397-1403.
- Kohno M, Yokokawa K, Horio T, Yasunari K, Murakawa K, et al. (1992) Atrial and brain natriuretic peptides inhibit the endothelin-1 secretory response to angiotensin II in porcine aorta. Circ Res 70: 241-247.
- Cheetham C, Collis J, O'Driscoll G, Stanton K, Taylor R, et al. (2000) Losartan, an angiotensin type 1 receptor antagonist, improves endothelial function in non-insulin-dependent diabetes. Journal of the American College of Cardiology 36: 1461-1466.
- Heitzer T, Krohn K, Albers S, Meinertz T (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 43: 1435-1438.
- Vehkavaara S, Yki-Järvinen H (2004) 3.5 years of insulin therapy with insulin glargine improves in vivo endothelial function in type 2 diabetes. Arteriosclerosis, Thrombosis, and Vascular Biology 24: 325-330.