Introduction
|
| Kaposi sarcoma (KS) is characterized by angioproliferative multifocal tumors of the skin, mucosa and less frequently the viscera. A close inspection of KS lesions reveals that they are largely comprised of cells of endothelial origin with a unique spindled morphology, and these are accompanied by a variable chronic inflammatory infiltrate. From an epidemiological standpoint, four different forms of KS are recognized: Classic (sporadic), African (endemic), acquired immune deficiency syndrome (AIDS)- associated (epidemic), and Transplant or immunosuppression associated (iatrogenic) KS (Table 1). Although these various forms of KS have different environmental and immunological components, the development of each depends upon prior infection with KSHV. However, KSHV infection alone is insufficient for the development of KS. For most cases of AIDS and Transplant KS it is well established that in addition to infection with KSHV, some form of immunodeficiency is also necessary for disease progression. However, several natural history studies indicate that additional factors may contribute to the development of KS in asymptomatic KSHV-infected persons. For example, not all AIDS patients develop KS even in the face of profound immunosuppression, only a minority of KSHV-infected transplant recipients develop iatrogenic KS, and people with classic or endemic KS are not typically immunosuppressed [1, 2]. The co-factors involved in classic and endemic KS have yet to be definitively elucidated. Various environmental and genetic factors have been implicated, as well as age, sex and malnutrition. The association of KS with the Koebner phenomenon, a condition where lesions initiate or recur at inflammatory sites of injury or trauma [3] and the recrudescent KS (KS flare) seen with the immune constitution inflammatory syndrome (IRIS) [4] suggest that inflammation plays a contributing role in oncogenesis. Progression of KS disease likely depends on a complex and as yet incompletely understood interplay between KSHV and the host immune system that allows for the establishment of a tumor-promoting environment. Like all herpesviruses, KSHV establishes a life-long infection in the host that depends upon |
 |
| virus-encoded immune evasion genes and genes that influence cellular proliferation, survival, migration, angiogenesis and cytokine/chemokine production. The host responds to persistent viral infection with its own chronic inflammatory response, thereby facilitating events that, particularly in the context of immunosuppression, could drive viral oncogenesis. Understanding the dynamic relationship between host and viral factors that drives KS oncogenesis is central to mounting effective strategies to prevent or ameliorate tumor development. In addition, deciphering KS pathogenesis will enhance our understanding of novel virus-induced mechanisms of tumorigenesis. There is a growing appreciation of the role that chronic inflammation plays in cancer development, and KS offers a unique opportunity to explore this topic. |
Historical Perspective
|
| In the late 19th century, Moritz Kaposi first described KS as a rare and relatively indolent disease of the lower extremities (classic KS). Throughout the 20th century, endemic KS was recognized in parts of Africa and the Middle East as a common cancer of children and adults. For example, in the pre-AIDS era, endemic KS in central Africa comprised up to 9% of malignant tumors in males [5]. Today, KS is the most prevalent malignancy among AIDS patients worldwide, and it has become the most common cancer in several sub-Saharan African countries where KSHV is endemic and HIV is widespread. Compared to the other forms of KS, AIDS-associated KS is an aggressive disease typically manifesting with disseminated and even visceral involvement, and has an untreated median survival of less than 2 years. KS occurs in AIDS patients who have not had highly active antiretroviral therapy (HAART) around 20,000x more often than in the general population [6]. In areas where highly active antiretroviral therapy (HAART) is widely available, there has been a dramatic decline in KS incidence. However, access to HAART is not universal, and this has led to a KS epidemic in many parts of Africa, along with the resultant potential for disease re-emergence elsewhere. KSHV-seropositive individuals with other forms of immunodeficiency (genetic, iatrogenic, idiopathic) are also at increased risk (~500x greater than the general population) for KS development [6]. In addition to KS, KSHV is also involved in two AIDS-associated B-cell cancers, primary effusion lymphoma (PEL), and the plasma cell variant of multicentric Castleman’s disease (MCD). KSHV seroprevalence varies geographically and demographically, and both sexual and non-sexual routes of transmission have been proposed. In the USA, prevalence ranges from approximately 5% among random blood donors to as high as 80% among groups of homosexual men [7]. In sub-Saharan Africa, where KS was endemic prior to the AIDS epidemic, seroprevalence ranges from 30-100% [8]. |
KSHV Biology
|
| KSHV is a g-herpesvirus in the Rhadinovirus genus encoding approximately 87 open reading frames (ORFs), 15 of which have no homologues in the other human herpesviruses. Like most herpesviruses, KSHV has two major modes of replication. In the lytic phase, entry, uncoating, and nuclear import are followed by a coordinated sequence of viral transcription, DNA replication, and assembly, followed by the final release of nascent virions. KSHV can also undergo a “latent” life cycle where only a small subset of viral genes is expressed. Here, after entry and translocation to the nucleus, the viral DNA circularizes, and multiple copies are maintained as episomes attached to the host chromosome via the viral latency associated nuclear antigen (Lana-1). Viral genomes are then replicated at roughly the same rate as the host chromosome, such that each daughter |
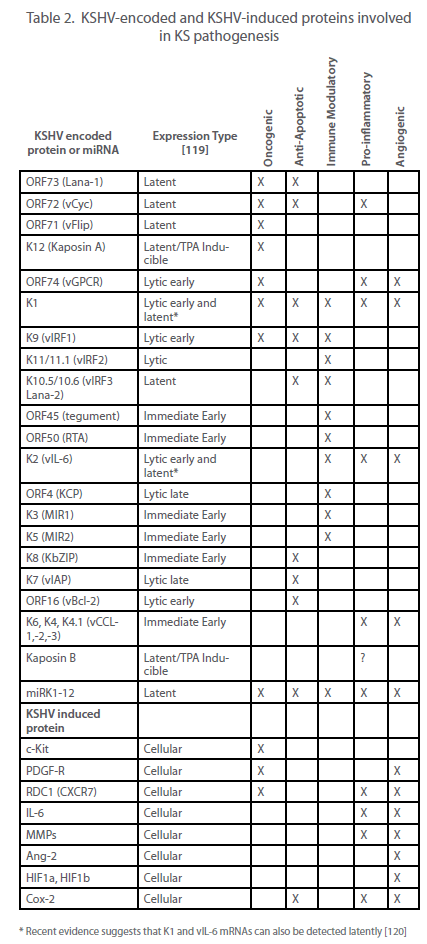 |
| cell receives several copies of the viral genome at cell division. Most cells in both KS lesions and KSHV-infected cultures are latently infected, with lytic replication occurring in only a subset of infected cells. However, as discussed elsewhere and summarized in Table 2, it appears that both phases of the virus life cycle play significant roles in the pathogenesis of KS. |
 |
KS Pathology
|
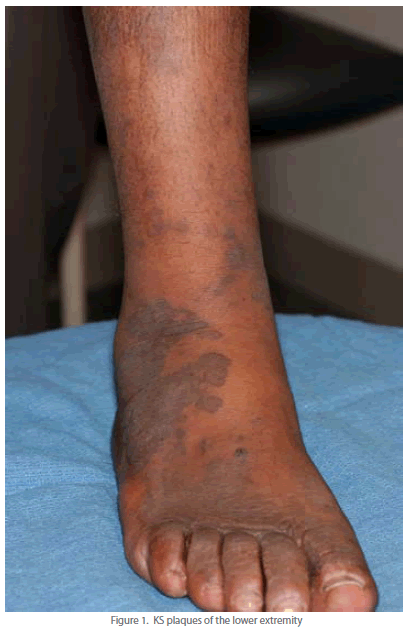
| Well-developed KS lesions (Figure 1) are comprised of spindledshaped tumor cells, abnormal vessels, and a variable chronic inflammatory infiltrate. Various histological variants (e.g. anaplastic KS, pyogenic-granuloma like KS, lymphangioma-like KS, amongst others) exist, that may mimic other vascular lesions [9, 10]. A premalignant stage of KS (so-called lymphedematous KS or KS in-situ) has been described in the setting of chronic lymphedema [11]. Chronic lymphedema results in local immune incompetence that, when coupled with lymphangiogenesis from developing collateral vessels, results in the development of KS lesions [12]. Depending on the inflammatory milieu and patient immune status, KS lesions may progress or regress. Progression evolves through distinct clinico-pathologic stages (Figure 2): they evolve from early (patch stage) flat macules into plaques (plaque stage) and subsequently nodular lesions (tumor stage). KS flare (so-called KS exacerbation) may occur as a result of IRIS following HAART, after corticosteroids, and with rituximab (chimeric monoclonal antibody against the protein CD20) therapy [13]. Regressing lesions are characterized by a partial or complete reduction of spindle cells accompanied by a prominent lymphocytic infiltrate [14]. The histologic appearance of KS is virtually identical in the different epidemiologic KS types. However, the clinical presentation varies among the different forms of KS (Table 1). Apart from molecular detection methods, KSHV can be identified and localized within KS lesional cells using immunohistochemistry. The commercial monoclonal antibody often used for this purpose is directed against the C-terminus of the latency-associated nuclear antigen (LNA-1 or LANA-1) encoded by ORF-73. LNA-1 immunoreactivity in KS cells usually appears as stippled nuclear staining. |
 |
Origin of KS lesion cells
|
| Unlike most tumors that arise from the expansion of a single transformed cell, the majority of KS tumors are polyclonal [15, 16], although a small percentage have been shown to be monoclonal [17]. The majority of tumor cells that comprise a KS lesion have a distinctive spindle morphology and express endothelial markers such as CD31 (also called PECAM-1 for platelet endothelial cell adhesion molecule) and CD34 [18]. In addition to spindle cells (SC), KS tumors contain other cell types such as T-cell lymphocytes, monocytes, macrophages, erythrocytes and dendritic cells [19, 20]. Over the past 15 years many stu- dies have been published in an attempt to pinpoint the origin of the SC. Initial histologic studies suggested that SC were of lymphatic endothelial cell (LEC) origin [21]. The majority of SC express LEC-specific markers such as LYVE-1, D2-40 (podoplanin), and VEGFR-3, but markers of blood vascular endothelial cells (BEC) such as factor VIII (von Willebrand factor) and PAL-E have also been identified [22-25]. In addition, KS lesions are not found in areas devoid of lymphatics, such as the brain [26]. Two independent groups used microarray analysis in an attempt to resolve this question. Using comparative transcriptomics, Wang et al. found that the gene expression profile of KS tissue more closely matched that of LEC, confirming the majority of the histology data [27]. They further compared the transcriptomes of KSHV infected and uninfected LEC and BEC, and discovered that KSHV infection resulted in a reprogramming, such that their gene profiles converged. Hong et al. performed a transcriptomic analysis of KSHV-infected BEC and likewise concluded that infected BEC were reprogrammed towards a lymphatic expression profile, as exemplified by induction of the LEC specific marker PROX1 [28]. KSHV-induced expression of the lymphangiogenic molecule VEGF-C has been implicated in the reprogramming of BEC to LEC [29]. In addition, the viral miRNAs, miR-K6 and miR-K11 have recently been implicated in reprogramming LEC by downregulating the transcription factor musculoaponeurotic fibrosarcoma oncogene homolog (MAF) [30]. An alternate explanation for an increased LEC phenotype in vivo is that infected LEC may have a proliferative advantage over BEC. Support for this notion comes from in vitro studies demonstrating that the KSHV copy number is higher in infected LEC than BEC [27]. Another possibility is that the originator of the KS spindle cell is an uncommitted endothelial progenitor cell (EPC) defined as being CD34+ and VEGFR2+. A recent study identified the presence of uninfected EPC in biopsies from all stages of KS development, suggesting an active recruitment of potential target cells [31]. In addition, circulating EPC were increased in peripheral blood of classic KS patients [32] and two independent studies report direct infection of CD146+ EPC isolated from the blood of classic KS patients [33, 34]. The mechanism of the SC precursor infection is also unclear. In addition to plasma virus, KSHV has been detected in peripheral blood mononuclear cells (PBMC) such as B cells, T cells and monocytes of persons with KS [35]. Thus, precursor SC may be primary targets or could be infected following contact with PBMC, possibly during extravasation. The fact that KSHV affects cellular markers used to identify LEC and BEC, and the possibility that KSHV may infect progenitors and influence lineage commitment, prevents a definitive conclusion about the cellular origin of KS. |
Mechanisms of KS tumorigenesis
|
| Cellular transformation is a multi-step process involving increased proliferation, apoptosis prevention, and autocrine signaling pathways. Evaluating the neoplastic nature of the KS SC has been problematic, primarily because these cells do not behave like typical cancer cells. For example, KS explant cells grown ex vivo still require serum and growth factors for maintenance and do not readily grow in soft agar or form tumors in nude mice [36]. The lack of specific chromosomal abnormalities in the spindle cells also suggests an atypical neoplasm [37]. Interestingly, upon passage in culture, cells derived from KS tissue lose the KSHV genome suggesting either limitations in the in vitro culture systems or the inability of the virus to confer a fully immortalized or transformed phenotype. In agreement, KSHV infection of primary endothelial cells in vitro does not readily lead to immortalization or transformation, with the exception of one study [38]. Taken together, the current data suggest that the KS tumor microenvironment must play a critical role in maintaining the KSHV transformed spindle cell in vivo. Interestingly, a recent study suggests that KSHV induces the Warburg effect (a common tumor phenotype involving increased glycolysis with a concomitant decrease in oxygen utilization), which may be responsible for maintaining the latently infected endothelial cells [39]. |
| As discussed above, some aspects of the immortalization/transformation pathway must be induced by KSHV, and others must be provided by additional environmental, genetic or immunological factors. An extensive and ongoing body of work has addressed the contribution made by KSHV-encoded proteins towards cellular transformation leading to the development of KS. Because of KSHV’s tendency towards latent gene expression and its ability to induce tumors in infected individuals, a great deal of focus has been placed upon the latent gene products as likely mediators of tumorigenesis (Table 3). |
| The role of the KSHV lytic cycle in KS tumorigenesis is also a matter of debate. Initial theories postulated that since only a small percentage of the tumor cells express lytic proteins and that the viral lytic cycle results in cell death, it was more likely that latent viral proteins would mediate cellular transformation. While anti-herpesviral drugs that block viral replication such as ganciclovir and foscarnet have been shown to effectively prevent KS development if given prophylactically, there are no data to support the efficacy of these drugs for the treatment of pre-existing tumors [40-42]. Therefore, while KSHV latent genes are essential for genome maintenance, lytic genes probably play an important role in driving tumorigenesis via direct as well as paracrine mechanisms. Despite the limited number of lytic gene products expressed in KS lesions, several lytic proteins have been found to have intrinsic immortalizing or transforming ability (Table 4). One major drawback of these studies is that the transforming function of individual KSHV genes is usually analyzed in over-expression systems, in the absence of other viral genes. Similarly, the majority of the animal assays were conducted in rodent models, which are not natural hosts for KSHV. Another important caveat is that when expressed alo-ne, lytic genes can activate many cellular genes, but during a normal viral infection, a host shutoff function mediated by the lytic SOX (shut off and exonuclease) gene (ORF37) prevents the expression of most cellular genes [43]. Taken as a whole, these data indicate that KSHV encodes many proteins that individually have the ability to immortalize/transform cells in vitro and in vivo, but how they all work together to perform this function in the context of a normal viral infection remains obscure. |
 |
 |
Virus-induced cellular oncogenes
|
| In addition to expressing its own potential oncogenes, KSHV induces the expression of a variety of cellular genes with transforming abilities. Analysis of gene expression profiles in KS tissue or EC infected with KSHV in vitro has identified several genes whose functions could conceivably contribute to KS tumorigenesis [44-50] . In order to verify their oncogenic potential, some of these genes have been further studied using over-expression and gene silencing techniques. Using these methods, the receptor tyrosine kinase c-Kit and the chemokine receptor CXCR7 (also RDC-1) have both been shown to play a role in the in vitro transformation of EC by KSHV [44, 45] [51]. c-Kit is an oncogene with an established role in other cancers [52], and CXCR7 has been shown to have oncogenic function [53]. SCF and SDF-1, the ligands for c-Kit and CXCR7, respectively, are both abundantly expressed in KS lesions, thus raising the possibility that proliferation-inducing autocrine or paracrine growth loops are established in KSHV-infected cells. A recent study implicates the KSHV-induced homeobox gene Pax2 as an additional factor in the transformation of human dermal endothelial cells [54]. |
| Immune evasion |
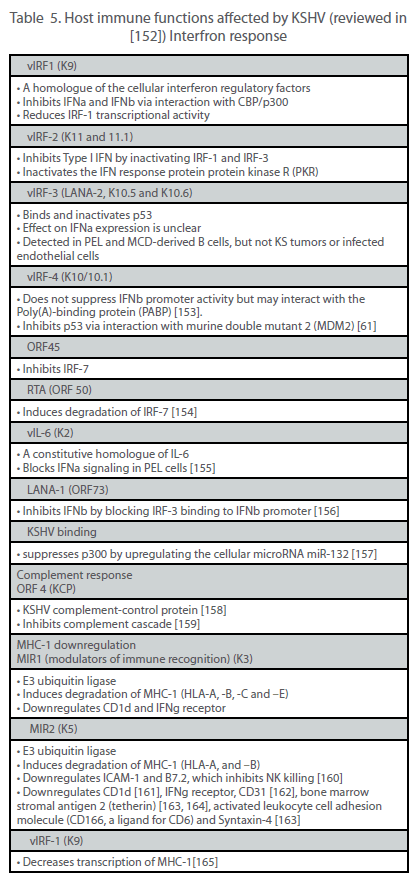
| Viruses have evolved a variety of mechanisms to evade the host immune system, thus allowing for efficient viral replication, dissemination and persistence. In the case of a tumor virus such as KSHV, the ability to escape immune detection might also enhance the oncogenic potential of the virus. The two main immune evasion strategies used by KSHV are the establishment of latency and the expression of immunomodulatory genes. During latency, the small number of viral genes expressed limits the number of epitopes that the immune system can target. By contrast, during the lytic cycle, more than 80 viral genes are expressed, potentially exposing the virus to immunodetection and subsequent clearance. However, KSHV encodes a number of proteins that actively hinder the innate and adaptive antiviral responses (Table 5). |
 |
| Apoptosis of infected cells is yet another mechanism employed by the host to combat viral infection, and KSHV has evolved mechanisms to counteract this as well. Because the prevention of premature cell death leads to enhanced cellular proliferation, the expression of anti-apoptotic genes might also contribute to viral immortalizing and transforming properties. KSHV expresses several lytic and latent proteins that block both extrinsic (FAS and TNFR-1 activated) and intrinsic (p53 activated) apoptosis pathways. vFLIP (latent) and K1 (lytic) can inhibit FAS-mediated apoptosis by blocking the activation of caspase 8 [55, 56]. KSHV also encodes several proteins that directly interact with p53 and inhibit p53-mediated apoptosis; Lana-1, vIRF-1, vIRF-3, RTA [57] and K8 (K-bZIP) [58]. K7 or inhibitor of apoptosis (vIAP) is a homologue of the cellular protein survivin [59] and appears to inhibit apoptosis by inducing p53 degradation [60]. Similarly, vIRF-4 has recently been shown to induce p53 degradation through its interaction with murine double mutant 2 (MDM2) [61]. KSHV also expresses a Bcl-2 homologue, vBcl-2 (ORF16) that inhibits p53-mediated apoptosis by blocking BAX [62]. Because the host cell dies from viral lysis, the lytic anti-apoptotic genes may only exert their effect long enough to maximize production of viral progeny. The latent anti-apoptotic genes would, however, ensure the survival of the cell containing the viral episome, thus resulting in long-term persistence of the virus. |
| The majority of immune evasion mechanisms utilized by KSHV are undertaken by lytic proteins, suggesting that their main purpose is to prevent eradication of cells producing viral progeny. Transient expression of genes such as K5 may also be crucial during both primary infection and initial establishment of latency. Alternatively, some of the lytic immune evasion genes might be expressed during latency, but at levels that are below the limit of experimental detection [63]. In this instance, lytic immune evasion molecules could play an active role in protecting the KS tumor from immune detection. The data reviewed thus far clearly illustrate that the virus has evolved a variety of mechanisms to combat the host antiviral response. However, like the latent gene products discussed above, most of these studies are performed in vitro, and frequently not in the context of the whole virus, thus making it unclear how effective these mechanisms are in vivo, or what role immune evasion plays in KS pathogenesis. |
Role of Chronic Inflammation
|
| Inflammation results primarily from the immune system’s response to infection or trauma. Normally, inflammation subsides once the infectious agent is cleared or the wound is healed, but in the case of persistent infections or uncontrolled cellular proliferation, inflammation can continue as long as the stimulus is present. In these instances the inflammatory process is no longer helpful, and instead becomes part of the problem. Many aspects of KS suggest that chronic inflammation associated with the lesion and/or viral infection plays a role in tumor pathogenesis. A role for inflammation in KS is exemplified by the association of KS with the Koebner (or isomorphic) phenomenon (KP). KP is broadly defined as induction by physical or other noxious insult of lesions of the same form (hence isomorphic) characteristic of the initial disease, or the disease to which the individual is predisposed [64]. A variety of cutaneous diseases demonstrate koebnerization. Several infectious agents, including human papilloma virus (HPV) and KSHV have also been associated with this phenomenon. While the types of trauma are varied (e.g., burn, bite, incision), the inflammatory response generated is thought to attract infected cells to the site as well as exacerbate the oncogenic properties of the viruses (reviewed in [3]). The recurrence of KS that sometimes accompanies IRIS (KS flare) is another example of an inflammatory response initiating or exacerbating KS (reviewed in [4]). |
| While it may seem counter-productive for a virus to encode proteins that both evade and activate the immune system (see Table 5), in the context of chronic inflammation it makes sense. Inflammation is primarily mediated by hyperactivation of the humoral arm of the immune system (Th2-mediated responses) and is often accompanied by a decrease in cellular immunity (Th1-mediated responses), which is the more effective antiviral response. Having finished our discussion of mechanisms used by KSHV to downregulate Th1-mediated responses such as suppression of IFNs and MHC-1 downregulation, we now focus on mechanisms that the virus uses to activate Th2 responses, leading to continued inflammation and KS tumor progression. |
| Pro-inflammatory cytokines |
| The type of immune response, be it Th1 or Th2, is regulated in part by the predominant chemokines present at the site of injury. KSHV encodes three chemokine ligands, vCCL-1 (K6), vCCL-2 (K4) and vCCL-3 (K4.1) that share homology with the cellular macrophage inflammatory protein MIP-1a__ (reviewed in [65]). These proteins bind to and signal through the G-coupled protein receptors CCR8, CCR3 and CCR4, respectively. These receptors are expressed on the surface of Th2 cells, and their activation leads to the preferential chemotaxis of Th2 lymphocytes to the site of infection. vCCL-2 can additionally act as an antagonist of CCR1 and CCR5, which are expressed on Th1 lymphocytes, thereby contributing to the evasion of the antiviral response [66, 67]. |
| Another KSHV protein that stimulates the proinflammatory immune response is vGPCR (ORF74). The constitutive activity of vGPCR induces myriad signaling pathways, including JNK/ SAPK, PLC/PKC, MAPK, and PI3K/Akt/mTor (reviewed in [68]. This leads to the activation of NF-kB, which has become increasingly linked to chronic inflammation and cancer progression, primarily via the prevention of apoptosis [69] , but also via the induction of angiogenesis [70]. The activation of multiple pathways by vGPCR in monocytes and T cells leads to the production of several proinflammatory cytokines including IL-1b, TNFa, IL-6, IL-2, IL-4 [71]. vGPCR-induced activation of NF-kB in endothelial cells results in expression of RANTES, IL-8 and GMCSF as well as the adhesion molecules VCAM-1, ICAM-1 and Eselectin [72]. Since vGPCR is a lytic gene product, any effect that its signaling has on tumor formation or progression probably occurs via a paracrine mechanism. KSHV also induces NF-kB via the expression of vFLIP, as described above. Because vFLIP is latent and therefore widely expressed in KS and PEL tissues, the activation of NF-kB is constitutive, not transient. This constant signaling from the tumor tissue could be a major stimulus for both proliferation and chronic infiltration by leukocytes. |
| Another source of KSHV-induced proinflammatory cytokines may come from the action of Kaposin B. Kaposin B is expressed from a CUG upstream of the K12/Kaposin A ORF, but is read from a different reading frame, such that Kaposin A is not expressed [73]. The mRNA that encodes the entire kaposin locus is abundant in KS tumor tissue, but the relative expression levels of the three kaposin isoforms is not clear [73]. A screen for cellular Kaposin B binding partners revealed that the MAPKassociated protein kinase MK2 was both bound and activated by Kaposin B. MK2 is normally activated by p38, leading to the stabilization of transcripts that harbor AU-rich elements (AREs) at their 3’ untranslated regions. There are thousands of such transcripts, including those that encode cytokines. Kaposin B was then demonstrated to stabilize the ARE-containing IL-6 and GM-CSF messages in KSHV-infected cells [74], suggesting that Kaposin B may contribute to the high levels of cytokines and potentially other chemotactic and angioproliferative factors observed in KS lesions. |
| Cellular IL-6 has long been associated with KS and MCD tumor development. It has been shown to enhance proliferation of KS cells in culture [36] and was found in high levels in blood samples from MCD patients, which correlated with disease progression [75]. The signaling pathways activated by IL-6 and vIL-6 that mediate an inflammatory response include JAK/STAT, MAPK, and PI3K/AKT/mTor [76-79]. Cyclooxygenase-2 (Cox-2) is another proinflammatory, pro-angiogenic molecule upregulated by KSHV, expressed in KS tissue and likely involved in KS pathogenesis [80]. |
| The importance of this pro-inflammatory component to KS tumorigenesis is highlighted by the success of the PI3K/AKT/ mTor pathway inhibitor rapamycin (sirolimus) in clinical trials for posttransplant KS [81-83]. |
| Angiogenesis |
| Angiogenesis is the formation of new blood vessels to increase blood supply to a site of injury, and is a normal inflammatory response that, when aberrantly stimulated, enhances tumor pathogenesis [84]. Two of the major cellular cytokines involved in angiogenesis are vascular endothelial growth factor (VEGF) and IL-6, which interact with their respective receptors to induce endothelial cell proliferation. IL-6 can also indirectly induce angiogenesis by upregulating VEGF [85], and KSHV vIL-6-expressing rodent cells and implanted tumors also induce VEGF [86]. KSHV encodes several other proteins that can induce VEGF, including all three KSHV-encoded chemokines (vCCLs). These have been shown to induce angiogenesis in experimental models [87, 88], and the mechanism for this activity is likely the induction of VEGF [89]. Similarly, vGPCR upregulates both VEGF and its receptor VEGF-R2 in KSHV-infected endothelial cells [90]. It has been postulated that this may create a paracrine feedback loop for continued cellular proliferation and angiogenesis [91]. Finally, K1 has also been demonstrated to induce VEGF expression and secretion [92]. |
| Matrix metalloproteinases (MMP) are key enzymes that remodel the extracellular matrix during tumor invasion, metastasis, and angiogenesis [93]. MMP-1, -2, -3, -9, and -19 are expressed in KS tumors [94, 95], and their importance to KS pathology has been demonstrated by the efficacy of an MMP inhibitor in treating AIDS-KS [96]. The KSHV protein K1 has been shown to induce the expression and activation of MMP-9, an MMP that is critical for the angiogenic switch in tumor progression [92, 97]. In vitro studies with KSHV-infected endothelial cells suggest that the virus induces MMP-1, -2 and -9 secretion and activation, thus allowing infected cells to invade an extracellular matrix [98]. A recent study indicates that KSHV, specifically LANA-1, may increase the levels of the extracellular matrix metalloproteinase inducer (emmprin) as a means to activate MMPs [99]. |
| Angiopoietins are another family of vascular growth factors important for angiogenesis. Angiopoietin-1 (Ang-1) binds and activates the receptor tyrosine kinase Tie-2, which synergizes with VEGF to promote endothelial cell proliferation and stabilization of blood vessels. In contrast, angiopoietin-2 (Ang-2) is an antagonist of Tie-2 that destabilizes existing vessels. Ang-2 is often upregulated at sites of vascular remodeling, while Ang-1 is ubiquitously expressed in endothelial cells. While both Ang-1 and Ang-2 are found in KS tumors, Ang-2 is expressed at higher levels [27, 100] and is induced in KSHV-infected endothelial cells [101]. |
| Tumor cells in environments that are low in oxygen induce angiogenesis via the stabilization of hypoxia-induced factors (HIF), which are transcription factors that interact with promoters containing hypoxia response elements (HRE). Two key regulators of angiogenesis, VEGF and VEGFR1, contain HREs [102]. Interestingly, KSHV encodes several means by which to upregulate HIFs. For example, vGPCR-dependent activation of both MAPK and p38 kinases leads to the subsequent phosphorylation and activation of HIF1a, and this is the likely mechanism for vGPCR’s induction of VEGF [103]. In addition, KSHV latent gene expression induces the transcriptional upregulation of both HIF1a and HIF1b [104]. Other important angiogenic proteins expressed in KS lesions are IL-1b, FGF-2 and PDGF-Rb__ (reviewed in [105]). The mechanisms for their upregulation are not yet known. |
| Most of the pro-inflammatory cytokines and angiogenic factors produced or induced by KSHV have likely evolved to create a highly proliferative environment that favors viral genome maintenance as well as the consistent, yet low level of reactivation from latency. Both lytic and latent genes are responsible, suggesting that both autocrine and paracrine mechanisms contribute to the microenvironment. In addition to virally induced factors, the persistent viral infection attracts a chronic leukocyte infiltrate, which in turn secretes cytokines, chemokines, enzymes, and growth factors that favor the growth of infected cells and contribute to KS progression. |
| Treatment |
| Treatment of KS is aimed at symptom palliation, tumor shrinkage, and prevention of disease progression [106]. Therapeutic decisions depend upon the presence and extent of symptomatic and extracutaneous KS, the HIV status of the patient and their corresponding HIV viral load, and the host immune status (e.g. CD4 cell count and co-morbid disease). The advent of HAART has lead to a marked reduction in the incidence, morbidity and mortality associated with AIDS-related KS [107]. Similar advances in chemotherapy and supportive care protocols have also allowed for KS to be more effectively managed. The recent identification of several molecular targets has provided several new potential therapeutic strategies [108][109]. |
| Most if not all patients with AIDS-related KS should receive antiretroviral therapy, assuming access to such therapy is available. Effective antiretroviral regimens are associated with both a reduction in the incidence of AIDS-related KS and a regression in size and number of existing lesions. The effects of HAART on KS are multifactorial and include inhibition of HIV replication, diminished production of the HIV-1 transactivating protein Tat, amelioration of the immune response against KSHV and perhaps some direct antiangiogenic activity of protease inhibitors. |
| The dramatic impact of HAART on KS is highlighted by a large Swiss cohort study of HIV-infected patients [110]. In this cohort, the relative risk (hazard ratio) of KS development between 1997 and 1998 (HAART-era) compared with a pre-HAART time period between 1992 and 1994 was 0.08 (95% confidence interval, 0.03- 0.22), representing a dramatic reduction in this AIDS-defining malignancy. Furthermore, it has been demonstrated that both protease inhibitor- and non-nucleoside reverse transcriptase inhibitor (NNRTI)-based HAART regimens are equally effective in protecting against KS. Immune reconstitution inflammatory syndrome, with paradoxical worsening of stable opportunistic infections and KS (KS flare), can occur in the setting of marked HAART-induced recovery of the immune system. |
| Systemic chemotherapy is given to patients with more advanced or rapidly progressive KS. Typical indications for such systemic therapy include widespread skin involvement, extensive KS of the oral cavity, symptomatic pedal or scrotal edema, symptomatic visceral involvement, and immune reconstitution inflammatory syndrome-induced KS flare. Although several chemotherapeutic agents (bleomycin, vinblastine, vincristine, hydroxydaunomycin, adriamycin, and etoposide) were shown to be active against KS in the past, liposomal anthracyclines (pegylated liposomal doxorubicin and liposomal daunorubicin) and taxanes (paclitaxel) constitute the backbone of current systemic cytotoxic therapy against KS. |
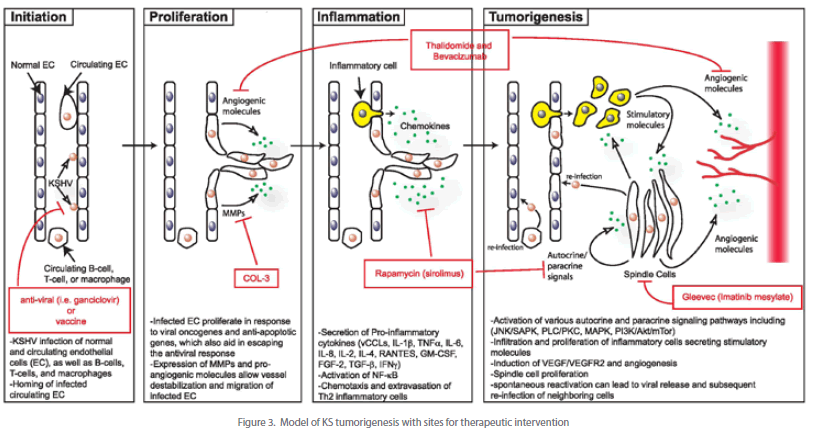
| Novel agents used to treat KS include angiogeneisis inhibitors, tyrosine kinase inhibitors, and matrix metalloproteinase inhibitors (see Figure 3). Thalidomide, which has significant antiangiogenic activity partly through inhibition of basic fibroblast growth factor-induced angiogenesis, has been shown to provide a partial response in treating KS [111]. Bevacizumab, a monoclonal antibody against VEGF, may provide similar potential activity in the treatment of KS. Imatinib mesylate, a PDGF-R and c-Kit inhibitor, has been found to result in marked clinical and histologic regression of KS [112]. The activity of other tyrosine kinase inhibitors (e.g. sorafenib and sunitinib) requires further investigation. Several trials have shown that COL-3, a chemically modified tetracycline that is a matrix metalloproteinase inhibitor, is beneficial in the treatment of AIDS-related KS [113]. Rapamycin (sirolimus), an inhibitor of the PI3K/Akt/mTor pathway, has been very effective in treating posttransplant [81-83] as well as classic [114] [115] KS and are currently being investigated for HIV-related KS. |
| There are no vaccines available for KSHV and research in this area has not been very active likely due to the lack of a suitable animal model and to the efficacy of HAART in dramatically lowering the incidence of KS. However, the prospects for a new non-human primate model that exhibits persistent KSHV infection with a robust anti-KSHV antibody response are promising [116]. A recent study using a recombinant murine herpesvirus 68 (MHV68) engineered to constitutively express the viral transcription activator, RTA protected mice from subsequent wildtype MHV68 challenge, potentially providing a novel vaccine approach for KSHV [117]. |
 |
Conclusion
|
| After initial KSHV infection, the development of KS depends upon the generation of a unique local microenvironment, as illustrated in Figure 3. This model, which is based on the data described in this review, depicts a potential pathway of KS development involving four stages of progression. These include the initiation of virus infection, proliferation and migration of infected endothelial cells, induction of a pro-inflammatory response and finally tumor formation. Key viral and cellular factors thought to play a role at each of these stages are indicated. In addition, current therapeutics and their targets are highlighted. |
| Thus far, much of the data regarding KS progression have come from analyses of the contribution made by individual KSHV genes. While such studies have provided invaluable insights into the tools available to the virus, the data have yet to definitively establish a hierarchy of events required for tumor progression. A more comprehensive view of the oncogenic process will greatly benefit from an animal model that can successfully recapitulate the disease and allow for the testing of defined viral mutants, in addition to rigorous clinical evaluation of asymptomatic and KS-afflicted KSHV-seropositive individuals. Only then will we gain a more complete understanding of the complex interplay between viral-encoded proteins and the host immune system, and how this interaction leads to KS. Fortunately, this interpretation of the disease as a convergence of immune evasion, oncogenesis, inflammation, and angiogenesis has broadened the potential targets for treatment. Ongoing clinical trials are evaluating inhibitors of VEGF, MMPs, and cytokine signaling pathways. As the role of additional molecular mechanisms involved in KS tumorigenesis, such as the Akt/mammalian target of rapamycin pathway and NF-kB, become apparent we can anticipate the development of new therapeutic agents to target these pathogenetic mechanisms. These mechanism-based agents are a superb example of therapeutic approaches bridging basic research with clinical practice. |
Acknowledgements
|
| The authors would like to thank the following agencies for their support. Ashlee Moses’ laboratory was supported by NIH grants CA 099906 and RR00163. Janet Douglas would like to acknowledge the Collins Medical Trust. |
| |
| |

