Keywords
Systemic lupus erythematosus; Bullous diseases; Teenagers
Introducción
El Lupus Eritematoso Sistémico Ampolloso (LESA) es una manifestación infrecuente del Lupus Eritematoso Sistémico (LES), el cual se incluye en el grupo de lesiones cutáneas inespecíficas. Esta patología tiene una incidencia de 0.2 casos por millón de habitantes y se presenta en menos del 5% de los pacientes con LES, afecta especialmente adultos jóvenes entre la segunda y cuarta década de la vida. Las mujeres, especialmente las de raza negra, son afectadas con mayor frecuencia que los hombres [1,2]. Es una enfermedad muy poco frecuente en niños, siendo más bien rara [3]. Se han establecido criterios para el diagnósticos de LESA por Camisa y Sharma, 1983 [1].
1. Diagnóstico de LES según los criterios de la American Rheumatism Association (ARA).
2. Vesículas y ampollas que surgen en la piel expuesta al sol, pero que no están limitadas a ella.
3. Histopatología compatible con Dermatitis Herpetiforme.
4. Inmunofluorescencia indirecta negativa para anticuerpos circulantes contra la zona de la membrana basal, utilizando la piel separada como sustrato.
5. Depósitos lineales o granulares de IgG, IgM o ambas y a menudo de IgA en la zona de la membrana basal, detectados por inmunofluorescencia directa de la piel de la lesión o de la piel de áreas sin lesiones.
Aparte de estos criterios diagnósticos, Gammon and Briggaman incluyeron la presencia de anticuerpos circulantes contra el colágeno tipo VII el cual es el principal componente de las fibrillas de anclaje de la unión dermoepidermica [4].
La presentación clínica de LESA es típicamente una erupción vesículo-ampollosa aguda generalizada. Puede aparecer en cualquier lugar del cuerpo, aunque existe predilección por la parte superior del tronco, el cuello, las regiones supraclaviculares, pliegues axilares, mucosa oral y vulvar [5], a pesar, que es una presentación rara de LES, y mas aún, en edad pediátrica, es importante que en la evaluación inicial esta sea considerada, de tal forma que se indiquen los estudios necesarios para establecer el diagnóstico específico.
Presentación de caso
Adolescente femenina de 14 años de edad, nacionalidad hondureña, procedente del área rural del departamento de Olancho, zona oriental del país, con historia de presentar ampollas de un mes de evolución, que inician en abdomen extendiéndose a miembros superiores y cara, las que posteriormente se generalizan en toda la superficie corporal, incluyendo palmas y plantas, acompañadas de dolor intenso y prurito leve. Al inicio la paciente fue tratada con medicamento tópico (desconocido) sin presentar mejoría, por lo que fue referida a un hospital terciario.
Al momento del ingreso, paciente en mal estado general con desnutrición aguda leve y desnutrición crónica grado II, se encuentra por debajo del percentil 3 según las curvas de crecimiento. Antropometría: peso: 25.7 kg, talla: 145 cm, constantes vitales: 90 latidos por minuto, 30 respiraciones por minuto, 35.7°C y Glasgow: 15/15. Examen cardiorrespiratorio sin hallazgos patológicos, abdomen sin visceromegalias y la piel mostraba ampollas tensas de tamaño variable, 0,5 hasta 2 centímetros con líquido claro en su interior, algunas denudadas y con formación de costra, también se observaban áreas hipo e hiperpigmentadas residuales, distribuidas en cara, tronco y extremidades, áreas denudadas en mucosa oral, genital, conjuntiva palpebral e inyección conjuntival, sin afección de piel cabelluda (Figura 1).
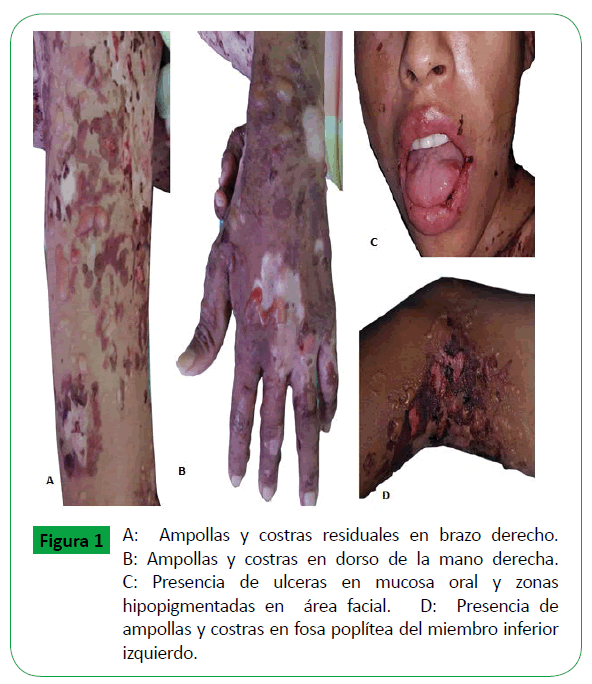
Figure 1: A: Ampollas y costras residuales en brazo derecho. B: Ampollas y costras en dorso de la mano derecha. C: Presencia de ulceras en mucosa oral y zonas hipopigmentadas en área facial. D: Presencia de ampollas y costras en fosa poplítea del miembro inferior izquierdo.
Como antecedentes personales se refería cuadro de pericarditis diagnosticada y confirmada por ecocardiograma de etiología no consignada, 5 meses atrás, manejada con prednisona y un ecocardiograma control que reportó resolución de la misma. No se constatan antecedentes familiares patológicos ni de enfermedades ampollares o sistémicas.
Exámenes complementarios
Hemograma: hemoglobina: 9.3 g/dL (12-18), glóbulos blancos: 10.43/mm3, (5.2-12.4), plaquetas 344/mm3 (130-400), linfocitos totales: 3.18/mm3, neutrófilos totales: 6.39/mm3. Examen General de Orina: cilindros granulares de 0 a 2 por campo. Proteinuria: 0.24 g/L en 24 horas, Química sanguínea: Creatinina: 0.7 mg/dl (0.6-1), Urea nitrogenada: 17 mg/dl (7-18), Proteínas totales 7.2 g/dL (6.4-8.2), Albumina 3.1 g/dL (3.4-5.0), relación albumina/globulina: 0.75 g/dL (1.5-1.9), TGO 49 u/L (15-37) TGP 31 U/L. Anti-DNA: positivo 1.933 U/ml (positivo mayor de 1.1).
Se tomó muestra de una ampolla para biopsia, la cual se mostró hiperqueratosis ortoqueratósica, estrato de Malpigio con discreta espongiosis y degeneración vacuolar de la capa basal, en la dermis formación de ampolla sub epidérmica e infiltrado inflamatorio con predominio de neutrófilos, dispuesto a nivel intersticial y perivascular (Figura 2). La muestra para Inmunofluorescencia directa se tomó de piel sana perilesional del glúteo, reveló depósitos granulares de IgA, IgG, IgM, fibrinógeno y C1q en la membrana basal de la epidermis, en correlación clínico, patológica y laboratorial son consistentes con el diagnóstico de LESA (Figura 3).
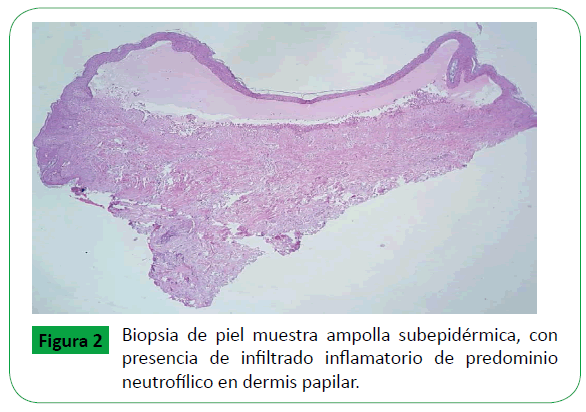
Figure 2: Biopsia de piel muestra ampolla subepidérmica, con presencia de infiltrado inflamatorio de predominio neutrofílico en dermis papilar.
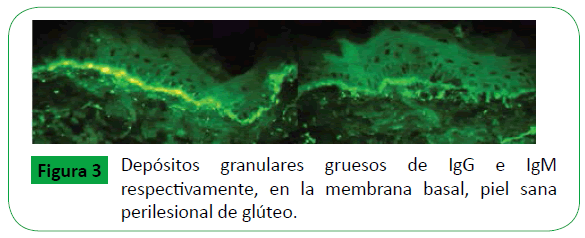
Figure 3: Depósitos granulares gruesos de IgG e IgM respectivamente, en la membrana basal, piel sana perilesional de glúteo.
Al inicio la paciente se trató como enfermedad vesículo ampollosa autoinmunitaria, recibiendo dosis única de Gammaglobulina 25 g (1 g/kg/día) e hidrocortisona (0.7 mg/kg/día), la paciente presentó mejoría transitoria, con recaída a los pocos días, por lo que se inició pulsos de Metilprednisolona 1 g cada 24 h por 3 días, mostrando una mejoría evidente en las erupciones y al confirmarse el diagnóstico de Lupus Eritematoso Sistémico Ampolloso, se continuó el manejo con Prednisona 25 mg por día (1 mg/kg/día), Azatioprina 50 mg (2 mg/kg/día) e Hidroxicloroquina 200 mg día, con mejoría clínica de las lesiones y posterior alta.
Discusión
El Lupus Eritematoso Sistémico (LES) raramente debuta con una manifestación ampollosa, siendo rara esta presentación en niños y adolescentes, en una revisión en PudMED, se encontraron reportados 13 casos en niños, los cuales estaban en un rango de edad de 8 a 18 años, 9 de ellos se presentaron en el sexo femenino y 4 casos en el sexo masculino. Cabe mencionar también que en la inmunofluorescencia directa el hallazgo más constante fue la presencia de IgG [6]. Adicionalmente se presenta una tabla donde se citan las principales características de algunos casos reportados desde los años 2010-2014 los cuales fueron utilizados como objeto de revisión para el reporte de este caso (Tabla 1).
| Autor y Año |
Género |
Edad |
Manifestaciones clínicas |
Características Histopatológicas |
| Saes-de-Oscaris et al. |
Femenino |
13 |
vesículas tensas y máculas de 3 días de evolución, localizada en cuello, tronco y región supraciliar izquierda, con base eritematosa, de 1-2 mm de diámetro |
biopsia: ampolla subepidérmica con marcada infiltración neutrofílica. |
| IFD*: depósitos granulares de IgG, IgA, C1q y C3 en la membrana basal |
| IFI*: no reporta |
| Saes-de-Oscaris et al. |
Femenino |
16 |
lesiones purpúricas y bullas de 7 días de evolución de 3-6 cm de diámetro con líquido claro en su interior localizada en tronco y extremidades |
Biopsia: atrofia de la epidermis con marcado queratinositos, necrosis, degeneración hidrópica de la membrana basal con formación de ampolla y edema de la papila dérmica. |
| IFD e IFI: no reporta |
| Caballero et al. |
Femenino |
10 |
ampollas tensas de contenido seroso y hemático de 21 días de evolución localizadas en cuello, tronco y posteriormente localizadas en toda la superficie corporal incluyendo pliegues y mucosas |
biopsia: ampolla subepidérmica con presencia de neutrófilos como célula predominante |
| IFD e IFI: no reporta |
| Santarcángelo et al. |
Femenino |
14 |
lesiones ampollares escasas y vesículas, con contenido claro en su interior, algunas erosionadas y con formación de costras, afectación de mucosa oral y linfadenopatía |
biopsia: infiltrado de neutrófilos en la zona de la membrana basal, fenómeno de leucocitoclasia, depósitos de fibrina y necrosis. |
| IFD: depósitos granulares de IgG, IgM y C3 |
| IFI: no reporta |
| Laurenco et al. |
Masculino |
10 |
vesículas desarrolladas sobre piel normal, afectando cara, mucosa oral y tronco |
biopsia: ampolla subepidérmica con microabscesos en la papila dérmica, infiltrado e inflamación perivascular. |
| IFD: depósitos de IgG, IgA e IgM. |
| IFI: no reporta |
| Laurenco et al. |
Femenino |
10 |
bullas y vesículas recurrentes en tronco de 2 meses de evolución, apareciendo posteriormente úlceras orales |
biopsia: bullas con infiltrado neutrofílico |
| IFD: depósitos de IgG, IgA, IgM y C3. |
| IFI: negativa. |
| Laurenco et al. |
Femenino |
5 |
vesículas y bullas en cuello y miembros inferiores, afectación de mucosa oral |
biopsia: ampolla subepidérmica con infiltrado de neutrófilos |
| IFD: depósitos de IgA, IgM, IgG |
| IFI: positiva |
* IFD: Inmunofluorescencia Directa ; * IFI: Inmunofluorescencia Indirecta
Tabla 1: Casos pediátricos de Lupus Eritematoso Sistémico Ampolloso.
Hasta el momento de la publicación, la paciente reúne 3 de los 4 criterios para diagnóstico de LES según la American Rheumatism Association (ARA), que es el primer criterio diagnóstico de LESA, constituidos por serositis (pericarditis), enfermedad renal (cilindros celulares en orina) y alteraciones inmunológicas (Anti DNA positivo). Si tomáramos en cuenta los nuevos criterios para LES propuestos por SLICC (sistemyc lupus international colaborating clinics) en 2012, los cuales presentan mayor sensibilidad que los criterios de ARA 1997, especialmente en la identificación precoz de pacientes con LES [7,8]; Como lo demuestra este caso, en el cual, además de los criterios arriba resumidos, se sumarían, hipocomplementemia a expensas de C3 y lupus cutáneo ampolloso. En estos nuevos criterios se requiere de 4 de los 17 criterios, incluyendo al menos 1 clínico y 1 inmunológico, ó nefritis lúpica con ANA o AntiDNA positivos, cumpliendo esta paciente de forma sobrada con criterios para LES. Para LESA, además este caso cumple con criterio clínico, histopatológico y los hallazgos en la inmunofluorescencia directa, con depósito de IgA, IgG, IgM y también C1q. No se realizó inmunofluorescencia indirecta.
En general el diagnóstico específico de las enfermedades ampollares es complejo y sólo es posible si se consideran particularidades como edad, sexo, características clínicas, histopatológicas e inmunopatológicas. En el caso que se presenta al inicio se consideró el diagnóstico clínico de Penfigoide Ampolloso (PA), el cual es la enfermedad ampollosa autoinmune más común pero afecta principalmente a individuos mayores de 60 años y es rara en niños. Clínicamente se presenta con ampollas grandes y tensas, puede estar precedido semanas a meses por una erupción pruriginosa y urticariforme, las lesiones se distribuyen principalmente en superficies flexoras de extremidades, cara interna de muslos, parte inferior de tronco, la afectación de mucosas es limitado en adultos, sin embargo en niños y adolescentes, la afectación de palmas, plantas y mucosas, suele ser la manifestación inicial [9].
A nivel histológico se caracterizan por la presencia de ampollas sub epidérmicas, pero se diferencian en el tipo de infiltrado inflamatorio que predomina, en PA los eosinófilos y en LESA los neutrófilos. En cuanto a los hallazgos en la inmunofluorescencia directa de piel perilesional, el PA muestra depósitos lineales de C3 y IgG, mientras que en el LESA se encuentran IgG, IgM o ambas y a menudo IgA [10,11].
Otro diagnósticos diferencial importante a considerar es la Dermatitis Herpetiforme (DH), que se presenta a cualquier edad, pero más frecuente en la tercera década de la vida y rara en la infancia, en adultos predomina en varones, mientras que en la infancia afecta principalmente al sexo femenino, se presenta con pápulas, placas, a veces con lesiones polimorfas de aspecto eccematoso o base urticariforme, sobre las cuales se desarrollan ampollas y vesículas, dejan pigmentación residual, las lesiones se distribuyen de manera simétrica, sobre todo en zonas de extensión como codos, rodillas, nalgas y hombros, característicamente muy pruriginosa, a diferencia de la paciente reportada, que presentaba lesiones sin un patrón de distribución característico, sin base eritematosa, ardorosas y con prurito leve. De forma excepcional la DH puede afectar mucosa oral y/o genital. Histológicamente se caracteriza por ampolla subepidérmica con infiltrado inflamatorio de neutrófilos, similar a LESA, y en la inmunofluorescencia directa se caracteriza por depósitos granulares de IgA en las papilas de?rmicas, en la membrana basal o en ambas [12].
La Epidermolisis Bullosa Adquirida (EBA), se asemeja con LESA en su fisiopatología, ya que ambas son caracterizadas por la producción de autoanticuerpos dirigidos contra el colágeno tipo VII de la unión dermoepidérmica [4]. En esta enfermedad, no existe predilección por raza y sexo, no obstante suele presentarse en la edad adulta y se caracteriza por ampollas tensas confinadas a áreas proclives a traumatismos sin eritema en la base o circundante y también involucra mucosas de forma limitada, pero cuando se presenta afecta mucosa oral, laríngea, esofágica y urogenital; al sanar dejan atrofia, alteraciones de pigmentación, lesiones cicatriciales y milia, lo cual no se observa en LESA. En la inmunofluorescencia directa de piel perilesional se observan depósitos lineales en la unión dermo-epidérmica de IgG en el 100% de los casos, habitualmente C3 y en proporción variable depósito de IgA e IgM [11,13].
La Dermatosis IgA lineal de la infancia, también conocida como dermatosis ampollar cro?nica benigna de la infancia, presenta lesiones vesiculo-ampollosas tensas, que aparecen sobre una base urticarial o piel sana, pueden adoptar morfología lineal o anular, rodeando áreas eritematosas dando el aspecto muy característico de "collar de perlas", “rosetas” o "racimos de joyas", también es característico la aparición de lesiones vesiculares en la periferia de lesiones antiguas, en forma de collarete, las lesiones tienden a la resolución espontánea sin dejar cicatriz y suelen ser pruriginosas, se describe una distribución clásica en cara (regio?n perioral), el abdomen, axilas, ingle y el perine?, de manera sime?trica o asime?trica, pero puede afectar todo el tegumento. El compromiso de mucosas es variable, afectando principalmente la cavidad oral, pero también puede haber compromiso genital, ocular y nasal. La histología muestra ampolla subepidérmica con infiltrado inflamatorio de neutrófilos y en cantidad variable de eosinófilos. En la IFD se caracteriza por el depósito lineal de IgA en la unión dermoepidérmica [14].
El tratamiento de primera elección para LESA se basa en el uso de Dapsona, una sulfona, que ha controlado efectivamente un número considerable de erupciones cutáneas, el mecanismo de acción de este medicamento es antiinflamatorio e inhibe las funciones de los leucocitos polimorfonucleares y la activación del complemento mediante la vía alternativa. Usualmente los pacientes tratados con Dapsona (2 mg/kg/dia) obtienen una mejoría considerable de la erupción, en 1 a 2 días hay disminución en la aparición de nuevas ampollas y posteriormente curación de lesiones ya existentes, sin embargo la mejoría de las erupciones no siempre significa mejoría de las manifestaciones sistémicas, incluso se ha reportado empeoramiento de las lesiones después de la administración de este medicamento [15]. En nuestra paciente no se utilizó porque en ese momento no había disponibilidad.
Los corticosteroides son usualmente requeridos para disminuir los síntomas clínicos, además de las anormalidades laboratoriales, siguen siendo un pilar para inducir remisión en pacientes con LES y son una terapia alternativa cuando los pacientes no responden a otro tipo de drogas en el tratamiento de LESA [15]. La paciente objeto de esta publicación recibió terapia con corticosteroides, al inicio por vía intravenosa luego se cambió a vía oral y el uso de estos evidencio mejoría clínica en la paciente después de la aplicación de dosis adecuadas.
Los antipalúdicos son útiles en especial para el manejo de las lesiones cutáneas, estos medicamentos espacian los brotes ampollares [16]. Al parecer los antipalúdicos tienen un efecto fotoprotector en los pacientes con lupus. Muchos autores han reportado una tasa de respuesta del 75 a 80% basados en experiencia clínica, generalmente son bien tolerados y no causan inmunosupresión sistémica, o toxicidad hepática y renal como la Dapsona [15]. En el caso al momento del diagnóstico definitivo se usó hidroxicloroquina 200 mg cada día observándose una notable respuesta en la desaparición de ampollas.
Otro medicamento utilizado en el manejo de esta paciente fue la Azatioprina, un profármaco, que al ser combinado con altas dosis de glucocorticoides es efectivo, una alternativa en aquellos pacientes con mala respuesta a la Dapsona, aunque la resolución de las lesiones es más lenta. Otras terapias utilizadas con buenos resultados son las inmunoglobulinas, además de Ciclofosfamida, Sulfapiridina y Metotrexate, la experiencia con estas drogas es limitada. También se ha informado respuesta exitosa al tratamiento con Mofetil-micofenolato y Rituximab en casos refractarios [1,17].
La paciente se está manejando por consulta externa, la Prednisona se disminuyó progresivamente hasta llegar a una dosis de 5 mg cada 2 días, Azatioprina 50 mg/día e hidroxiloroquina 200 mg/día. Estos medicamentos al usarse de manera combinada fueron fundamentales en el tratamiento de esta paciente y en la posterior resolución de las lesiones. A 2 meses después del egreso permanece sin lesiones, no quedaron cicatrices y desaparecieron los cambios pigmentarios en piel. Así mismo se comprobó una mejoría de estado nutricional, peso 30.5 kg obteniendo una ganancia de 5.2 kg en relación al peso durante el inicio de la enfermedad.
Al tratarse de una enfermedad poco frecuente en la población general y muy rara en niños consideramos importante incluir el diagnóstico de LESA en el diagnóstico diferencial de las enfermedades ampollares. El principal diagnóstico diferencial en población adulta es el Penfigoide pero en niños es la Dermatitis IgA lineal. Histológicamente el LESA es indistinguible de la Dermatitis Herpetiforme, tal como se menciona en los criterios diagnósticos de esta enfermedad.
Conflicto de Interés
Los autores declaran no tener ningún conflicto de interés.
17606
References
- González L, Vásquez G, Restrepo M (2012) Enfermedad cutánea ampollosa en el lupus eritematoso sistémico. Iatreia 25: 229-239.
- Caballero R, Morel Z, Gutiérrez O, Martínez G, Di Martino B (2011) Lupus Eritematoso Sistémico Bulloso en una niña de 10 años de edad. Reporte de un caso. Pediatr 38: 126-129.
- Bielsa I, Rodríguez C (2010) Manifestaciones cutáneas del lupus eritematoso. Inmunología 29: 100-110.
- Lourenco DMR, Gomes RC, Aikawa N, Campos L, Romiti R, et al. (2014) Childhood-onset bullous systemic lupus erythematosus. Case report. Lupus 23: 1422-1425.
- Sebaratnam DF, Murrell DF (2011) Bullous Systemic Lupus Erythematosus. Dermatol Clin 29: 649-653.
- Saez de Oscaris M, Espinoza-Rosales F, López-Corella E, Leon-Bojorque E (2010) Bullous lesions as a manifestation of systemic lupus erythematosus in two Mexican teenagers. Pediatr Rheumatol 8:19.
- Ungsprasert P, Sagar V, Crowson CS, Amin S, Makol A, et al. (2016) Incidence of systemic lupus erythematous in a populatio-based cohort using revised 1997 American College of Reheumatology and the 2012 Systemic Lupus International Collaborating Clinics classficatio crtiteria. Lupus.
- Larosa M, Iaccarino L, Gatto M, Punzi L y Doria A (2016) Advances in the diagnosis and classification of systemic lupus erythematosus. Expert Rev Clin Immunol 8: 1-12.
- Hurtado-Montiel VS, Morales-Trujillo ML, Moreno-Collado CA (2008) Penfigoide ampolloso en la infancia: comunicación de un caso y revisión bibliográfica. Dermatología Rev Mex 52: 38-42.
- Sami N (2011) Approach to the patient with autoimmune mucocutaneous blistering diseases. Dermatol Ther 24: 173-186.
- Sainz De la Maza Serra MT (2004) Síndromes mucocutaneos. En: Benitez del Castillo JM, Durán de la Colina JA, Rodríguez Ares MT. Superficie Ocular. España: Sociedad española de oftalmología pp: 187-199.
- Iranzo-Fernández P (2010) Dermatitis herpetiforme: Patogenia, diagnóstico y tratamiento. Med Cutan Iber Lat Am 38: 5-15.
- Sucar Batista M, Serrano García Y, Miranda Vergara T (2015) Epidermolisis bullosa adquirida. Reporte de caso. Revista Electrónica Dr. Zoilo E. Marinello Vidaurreta 41.
- Fuentelsaz del Barrio V, Campos Domínguez M (2013) Dermatosis IgA lineal de la infancia. Rev Pediatr Aten Primaria 15: 141-145.
- Duan L, Chen L, Zhong S, Wang Y, Huang Y, et al. (2015) Treatment of Bullous Systemic Lupus Erythematosus. J Immunol Res 2015: 167064.
- Santarcángelo S, Navacchia D, Cao G, Valle LE (2012) Lupus eritematoso sistémico ampollar. Presentación de un caso pediátrico. Rev Asoc Med Argent 125: 34-35.
- Hansen CB, Dahle KW (2012) Cutaneous lupus erythematosus. Dermatol Ther 25: 99-111.

