Keywords
Systemic Lupus Erythematosus, Autoantibodies, Immunophatogeny.
Introducción
El fenómeno de autoinmunidad fue descrito en 1899 por Paul Ehrlich [1], quien describe que las enfermedades autoinmunes se conocen por presentar una supresión de la tolerancia del sistema inmunológico a antígenos propios, es decir, el desarrollo de una enfermedad causada por una reacción de autoagresión [1,2,3].
El Lupus Eritematoso Generalizado (LEG), es una enfermedad autoinmune [4], inflamatoria, crónica [5] y multisistémica [6,7], cuya etiología es multifactorial [8,9] y es propia del tejido conectivo [10]. El LEG se caracteriza por la producción de autoanticuerpos [1] y puede ser una enfermedad órganoespecifica o sistémica, lo que puede afectar a uno o varios órganos, generando varias manifestaciones clínicas debido a la ubicuidad de estos autoantígenos [11].
Afecta al 0.1% de la población y es más frecuente en mujeres que en hombres con relación 9:1 [10,11,12]. Otros autores mencionan que la relación es de 6:1 [13]. En 1954 la tasa de supervivencia era del 50%, actualmente es del 97%. Por otra parte, se ha descrito que cerca del 25% de personas con LEG también pueden desarrollar el síndrome de Sjörgren [5].
En la patogenia están involucrados factores ambientales, genéticos [14] y hormonales [5,15]. El LEG se ha reportado en todo el mundo, aunque estudios demuestran que su prevalencia es mayor en Europa, América y Asia en comparación con Australia y África, lo que indica que ciertas razas tienen más susceptibilidad genética para el desarrollo de la enfermedad, como es el caso de la ascendencia africana que vive en los Estados Unidos, las Islas del Caribe, el Reino Unido y Europa [13]. Otros autores mencionan que aparte de la ascendencia africana, los hispanos y asiáticos son más susceptibles en pacientes con ancestros caucásicos [16]. Estudios recientes hacen una comparación entre pacientes del norte de España y pacientes hispanoamericanos de Texas cuyos ancestros son mexicanos mostrando que el LEG en los hispanoamericanos es más severo [15].
Factores de riesgo en LEG
Los factores de riesgo que participan en el desarrollo de la patogenia son ambientales, genéticos y hormonales que contribuyen a la perdida de la tolerancia inmunológica [13,11]. Un individuo genéticamente susceptible necesita la exposición de múltiples estímulos ambientales que condicionaran el desarrollo de la enfermedad. Los alelos asociados a la enfermedad, están presentes en personas sanas, pero cuando se encuentran en forma combinada y expuestos a estímulos estresantes como la radiación ultravioleta (Figura 1), virus, metales pesados, fármacos como hidralazina, procainamida, isoniacida, clorpromacina, metildopa y minociclina (Lupus inducido por drogas), entre otros, desencadena el fenotipo del Lupus [5,11,17].
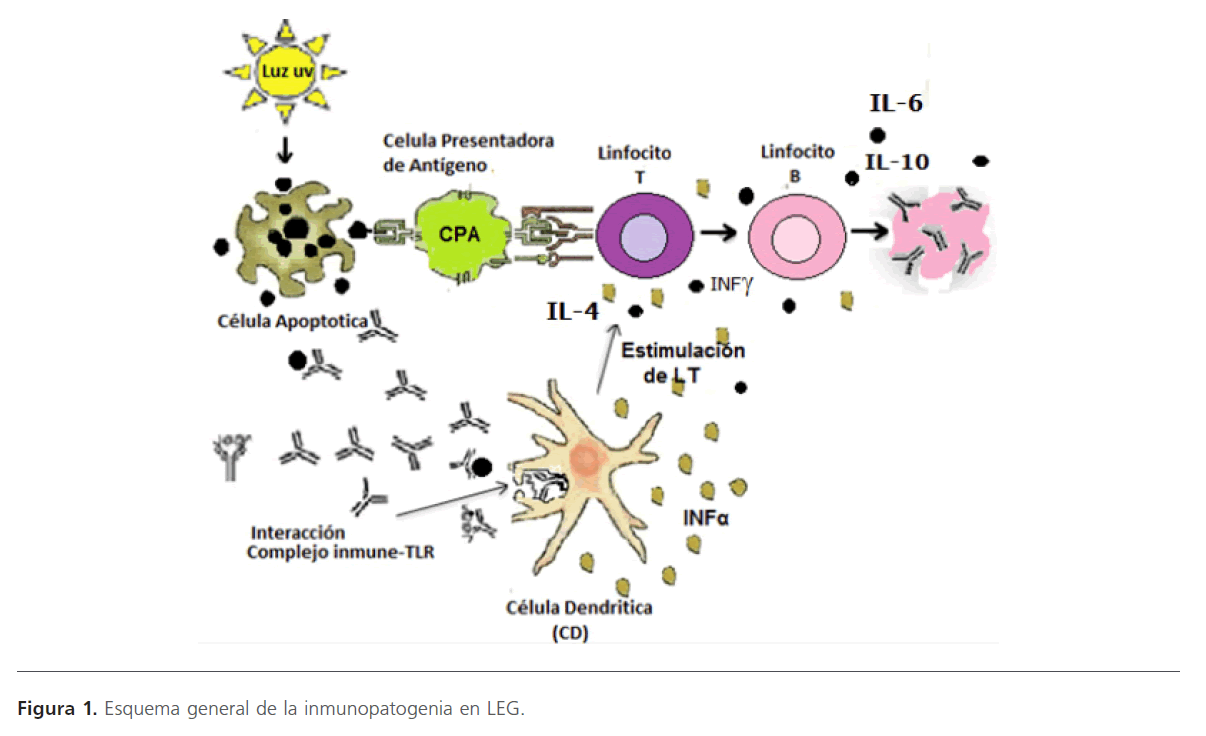
Figura 1: Esquema general de la inmunopatogenia en LEG.
Las hormonas juegan un papel importante en LEG, ya que se ha observado su mayor incidencia en mujeres de edad reproductiva, donde se observa una agresión mayor durante los ciclos menstruales, en los periodos post-parto y en la gestación [13,17]. Los estrógenos causan la pérdida de la tolerancia y facilitan la sobrevida de los linfocitos B autorreactivos a través del aumento en la expresión génica y de los linfocitos T CD40. Estas hormonas in vitro, actúan en la reducción de apoptosis, de células mononucleares de sangre periférica, reducción de niveles de TNF-a, activan las células dendríticas (DC) y reducen el número de colonias de granulocitos y macrófagos.
Otra hormona es la prolactina (PRL), donde ciertos estudios demuestran su papel como una verdadera citocina, y su expresión parece estar asociada a la clínica del LEG [17], y al igual que los estrógenos, estimula la expresión de linfocitos T CD40 y el rescate de linfocitos B autorreactivos [11].
Aunque la mayor incidencia es de mujeres en edad fértil, también se presenta en niños, mujeres postmenopáusicas y en hombres, donde es más severa la enfermedad [11,13]. También se ha sugerido que la edad en la que se desarrolla la enfermedad tiene un efecto modificador en su presentación, es decir, si se presenta en una edad avanzada es menos probable la afectación a los principales órganos y sistemas. El LEG es más frecuente en niños y mujeres jóvenes, pero en la edad pediátrica, la severidad no está influida por el género, como se presenta en los adultos [13].
Inmunopatogenia de LEG
El LEG se caracteriza por la activación e hiperreactividad de LB [18] y formación de autoanticuerpos [19], mediados por la secreción de diversas citocinas producidas por linfocitos T (LT) [6,20]. Los principales indicadores de la enfermedad son los autoanticuerpos, complejos inmunes, factores del complemento y las células autorreactivas [5]. El LEG incluye además inflamación e incremento de muerte celular por apoptosis, donde se presenta una deficiencia en la eliminación de restos celulares o cuerpos apoptóticos por los fagocitos, cuyos restos se transportan en vesículas para ser liberados, obteniendo una generación constante de autoantígenos modificados (que en un individuo sano el sistema fagocítico las degrada antes de su liberación), exponiéndolos al sistema inmune como se muestra en la figura 1. Lo anterior lleva a la generación de autoanticuerpos que están dirigidos a antígenos propios [22]. Los autoanticuerpos se unen a los antígenos propios (RNA,

Figura 1: Manifestaciones clínicas: vasculitis.
(Imagen aportada por el Dr. Martín Zapata Zúñiga).
DNA, restos apotóticas, etc.) que entran al torrente sanguíneo. A estas uniones se le denomina complejos inmunes (antígeno-anticuerpo) como se aprecia en la figura 1 [21], los cuales se pueden depositar en las membranas basales llevando a la activación del complemento [22], lo que provoca la aparición del proceso inflamatorio y en consecuencia manifestaciones clínicas dependiendo del órgano blanco [10], un ejemplo son aquellos complejos inmunes formados por anticuerpos anti-DNA de doble cadena (anti-sdDNA) que participan en el daño renal y cutáneo [21].
Otros complejos inmunes se unen al receptor FcyRIIa de las células dendríticas plasmocitoides (DCp) o al receptor de antígeno del LB específico. Ya acoplados al receptor, serán internalizados y se unirán a uno de los receptores de la membrana endosómica Toll like receptor (TLR) que son parte del sistema inmunitario innato. Se han descubierto 13 TLRs distintos localizados en membrana celular o en vesículas endosómicas y tienen gran especificidad por su ligando respectivo. El TLR-7 reconoce específicamente RNA de cadena simple y el TLR-9 a DNA (ambos en membrana endosómica), ya unidos éstos complejos antígeno-anticuerpo con los receptores TLRs activan una cascada de cinasas que conllevan a la síntesis masiva de interferón alfa (INF-a), llamado interferón tipo 1 (Figura 1). También se han identificado estímulos endógenos capaces de inducir interferón tipo 1 por vías independientes a los TLRs. Además de la estimulación de INF-a, se producen las citocinas IL-2, IFNg, IL-4, IL-6, IL-10 y factor del crecimiento transformante beta (TGF–9), BAFF (BLyS) y APRIL, estas dos últimas promotoras de la supervivencia y diferenciación de LB [3]. En la figura 1 se muestra la producción y efecto de citocinas IL-4, IL-6 e IL-10.
Después de la síntesis del interferón tipo 1, se estimula la activación de los LT (células autorreactivas que escaparon a la tolerancia central), los cuales interaccionan con los LB, donde ocurre la unión de receptores de los linfocitos LT (TCR) con el péptido antigénico a través del complejo mayor de histocompatibilidad (CMH), produciéndose el acoplamiento entre las moléculas CD40 de LB como célula presentadora de antígenos y su enlace CD40L de los LT. Este acoplamiento de los LT, induce la producción de citocinas, las cuales al actuar sobre los LB estimulan la producción de anticuerpos [10] (Figura 1), llevando a una mayor formación de complejos inmunes y la activación de LT citotóxicos (LTc) (liberan ácidos nucleicos y proteínas por mecanismos de citotoxicidad dependientes de granzimas) que entregan autoantígenos y continúan el circulo patogénico [3,22].
Citocinas
Una de las citocinas más importantes en la patogenia del LEG, es el interferón tipo 1, el cual, actúa como una molécula de estrés en el sistema inmune, señalando peligro en la patogenicidad y sus efectos influyen sobre la mayoría de los procesos de regulación, es decir, actúa como un factor inmunomodulador en varias células blanco, como células dendríticas (CD), CDp, LTc, células natural killer (NK), LT cooperadores (Th), LB, etc. [21]
Las IL-4, IL-6, e IL-10, ayudan al desequilibrio de las Th1 y Th2, favoreciendo la respuesta Th2, las cuales tiene una participación con INFg al inicio de la enfermedad, favoreciendo Th1 y Th2 en fases tardías y finalmente estas interleucinas participan también con TGF-β coestimulando las células T nativas, activándolas para diferenciarse a células Th1 o TH2.
Para la activación constante de LB, encontramos el factor activador de LB (BAFF), el cual es regulado por INFg, IL-10 e IL-6. El INFg también participa en el desequilibrio de la tolerancia hacia autoantígenos, aumentando la expresión de MHC de clase II. Otra citocina descrita en LEG, es el TNFa, que junto con IFNg e IL-10 están relacionadas con nefritis lúpica y la IL-6 con lupus discoide cutáneo [23].
Manifestaciones Clínicas
Los pacientes manifiestan alteraciones en el sistema nervioso central (SNC), renales, articulares, hematológicas, vasculitis [5] (Figura 1), arteriosclerosis, arteriopatía coronaria y osteoporosis entre las más frecuentes [24]. Se cree que es la enfermedad autoinmune con más variedad serológica y clínica [25] y es por ello que se considera como no órgano-específica, ya que puede afectar a cualquier órgano de muy diversas formas y grados, por consiguiente presenta un número amplio de autoanticuerpos en cada paciente [26]. La aparición de síntomas generales más frecuentes de la enfermedad son: a) Astenia (80-100 %), presente en los periodos de actividad de la enfermedad; b) Pérdida de peso (60 %): suele ser inferior al 10 %, y c) Fiebre (80-97 %): es un síntoma de actividad de la enfermedad [10, 17, 27].
Autoanticuerpos
En el LEG se han encontrado anticuerpos contra muchos autoantígenos (nucleares, de membrana celular, proteínas plasmáticas y de la matriz extracelular), pero la mayoría se unen a antígenos nucleares [22].
Los anticuerpos antinucleares (ANA) se presentan en un 98- 99.5% en pacientes con LEG, el 0.5% que no los presentan se denominan seronegativos. Los anticuerpos son un factor característico, pero no sólo se presentan en esta patología, inclusive se encuentra presente en títulos bajos en un 5% de la población en general aumentando su prevalencia con la edad [16,22]. Se pueden encontrar 3 tipos de ANA, el primer tipo se conoce como ANA naturales y son los encontrados en títulos bajos en el organismo, el segundo se produce como el resultado de procesos infecciosos y sus títulos bajan cuando se resuelve el proceso de infección que les dio origen, y el tercer tipo de ANA son los autoinmunes, los cuales reflejan la pérdida de tolerancia a lo propio y su origen es multifactorial [28].
El estudio de los ANA se inició con la identificación de células LE en pacientes con LEG, descrito por Hagraves en 1948, a partir de su descubrimiento estas células fueron utilizadas para confirmar el diagnóstico de LEG. Sin embargo años después se demostró su baja especificidad al encontrarlas presentes en Artritis Reumatoide con 25%, síndrome de Sjógren 15–20%, cirrosis pancreática (33%), hepatitis crónica activa 50–70% y en otras enfermedades 1–2% como miastenia gravis y púrpura trombocitopenia idiopática. En 1959 Holman demostró que las células LE se debían a la presencia de anticuerpos que reconocían antígenos nucleares [28,22].
A la fecha se conocen más de 100 antígenos específicos de LEG, sin embargo los más estudiados y con mayor especificidad son: DNA de cadena doble (dsDNA), DNA de cadena simple (ssDNA), antígenos nucleares extractables (ENA) como (Sm RNP, Ro y La), histonas y cromatina [13,22,28,29] (Tabla 1).
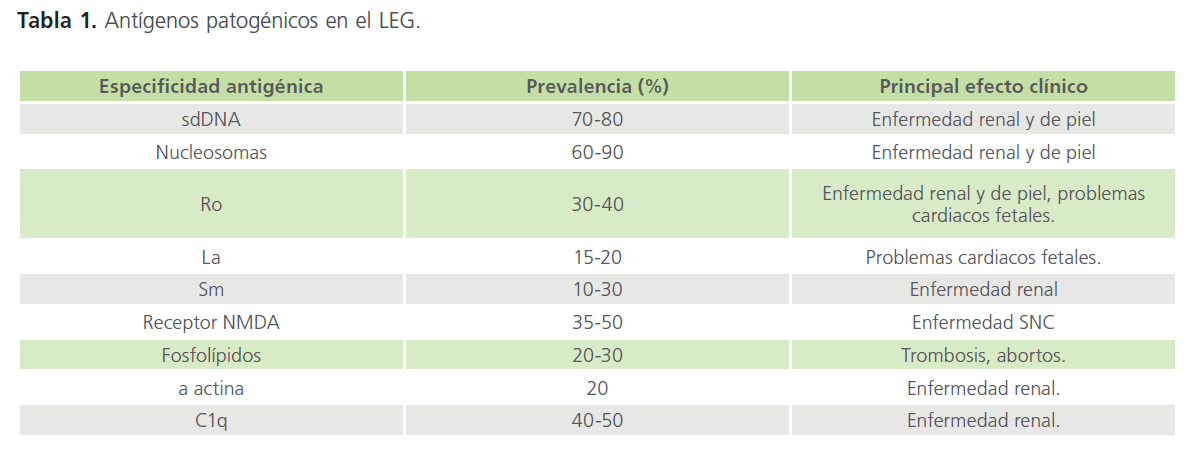
Tabla 1: Antígenos patogénicos en el LEG.
Los anticuerpo anti-sdDNA nativos o de doble cadena son muy importantes ya que son los principales para diagnóstico y análisis de la enfermedad, se observa que están presentes en el 40-60% de los pacientes, y es muy específica para LEG, en comparación con los anticuerpos anti-ssDNA que a pesar de que se halla en un 90% de los pacientes con LEG, es menos especifico ya que se encuentra en otras enfermedades autoinmunes.
Los antígenos DNA son los principales promotores en el daño renal, se encuentran ubicados en el colágeno de la membrana basal del riñón y la piel, donde los anticuerpos anti-DNA se unirán a ellos, desencadenando una respuesta inflamatoria donde interviene los múltiples mediadores inflamatorios [24].
Pruebas de laboratorio
Para demostrar la existencia de anticuerpos antinucleares [29] e identificación de antígenos como herramienta diagnóstica, se utilizan las técnicas de inmunoprecipitación y análisis de proteínas y de ácidos nucleicos, enzimoinmunoanalisis (ELISA) con extractos purificados o proteínas recombinantes, [3, 30] Western blot e inmunoflorescencia indirecta (IFI) [3,28,31]. Se comienza la identificación con la prueba de ANA por IFI y si ésta resulta positiva debe hacerse la confirmación mediante otras técnicas como ELISA y Western Blot. La técnica más reciente en LEG, es la inmunoaglutinación, la cual se basa en la aplicación de nano partículas de oro sensibilizadas con desoxiribonucleoproteínas que detectan los marcadores de LEG (anticuerpos específicos), siendo una técnica con alta sensibilidad diagnóstica [32].
Criterios de clasificación
El LEG se apoya en 11 criterios de clasificación para poder analizar los grupos de pacientes en estudios clínicos, de los cuales se necesita tener ≥ 4 criterios [5,33]. Estos criterios fueron descritos por el Colegio Americano de Reumatología (CAR) revisados en 1982 y modificados en 1997 [13].Un de los criterios son las ulceras orales (Tabla 2, Imagen 2).

Imágenes 2: Manifestaciones clínicas: Ulceras bucales. (Imagen aportada por el Dr. Martín Zapata Zúñiga).
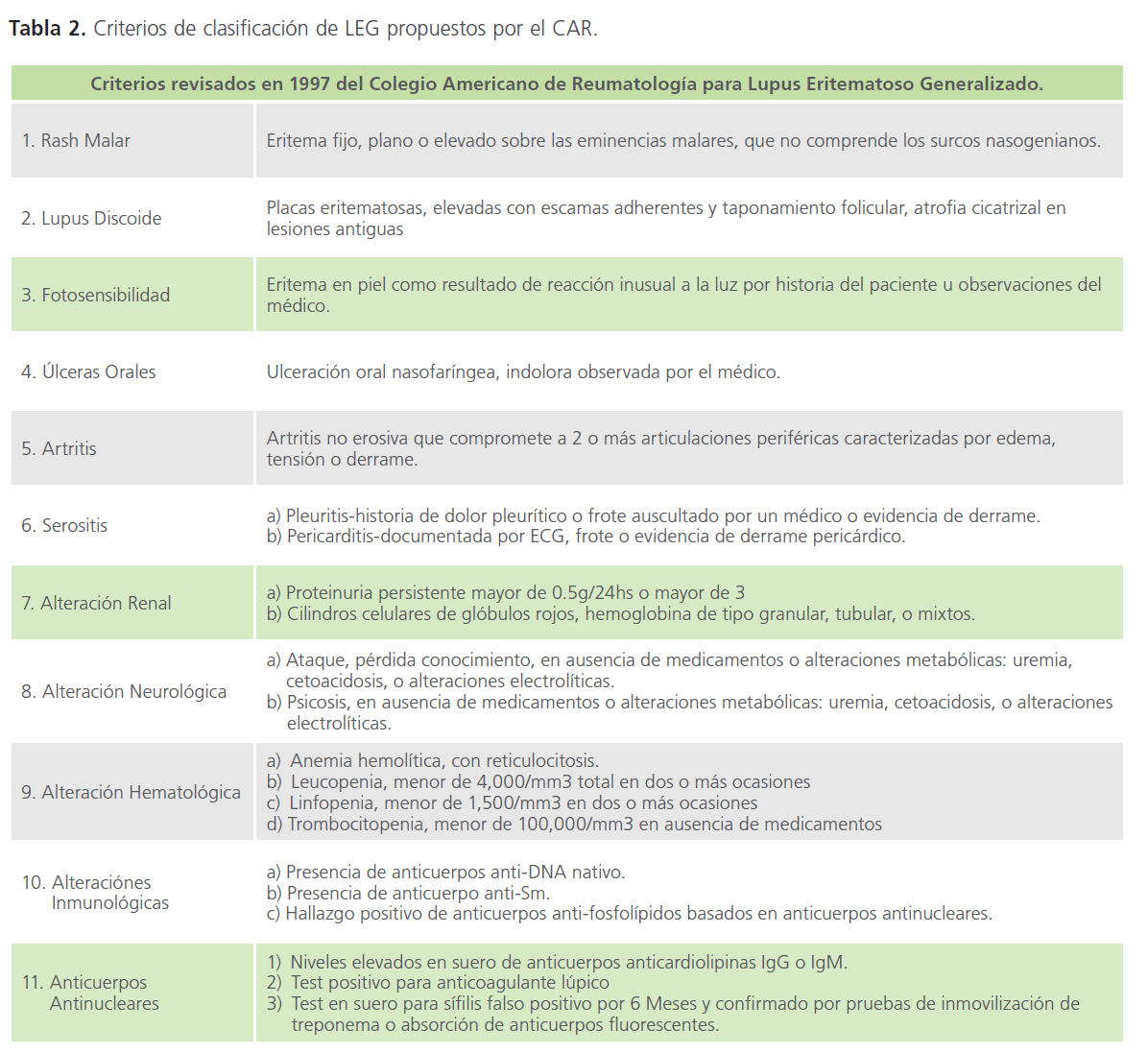
Tabla 2: Criterios de clasificación de LEG propuestos por el CAR.
Es importante mencionar que estos criterios no son diagnóstico, son de clasificación [5,34,35].
Conclusión
El Lupus Eritematoso Generalizado es una enfermedad autoinmune, con etiología desconocida, su patogenia es reconocida por la pérdida de la tolerancia, donde la autoagresión es generada por los autoanticuerpos ya que reconocen varios antígenos celulares. Esta patología se presenta más en mujeres que en hombres con una relación 6-9:1, aunque también está presente en niños y mujeres post-menopaúsicas. Hay factores que condicionan el desarrollo de la enfermedad que son hormonales, ambientales y genéticos. Esta enfermedad ha generado diversos estudios a lo largo del mundo, tratando de entender la compleja etiología y su desarrollo, pero hasta el día de hoy a pesar de los avances tecnológicos y nuevos descubrimientos sobre esta patología todavía queda mucho por saber y entender.
700
References
- Mahler M, Fritzler MJ. Epitope specificity and significance in systemic autoimmune diseases. Ann. N.Y. Acad. 2010 Sci. ISSN 0077-8923 267–287.
- Jara LJ. La interacción inmuno-neuro-endocrina en enfermedades reumáticas autoinmunes: un nuevo desafío para el reumatólogo. Reumatol Clin. 2011;7(2):85–87
- Gelpí SC. Anticuerpos en las enfermedades autoinmunitarias sistémicas. Especial mención al lupus eritematoso sistémico. ReumatolClin. 2008;4 Supl 1:S11-6
- Marenco JL, Fernandez-Nebro A. Experiencia con rituximab en el tratamiento de pacientes con lupus. Reumatol Clin.2010;6(S2):28–33
- Ramirez G, Gamarra G, Badillo AR, Daza BN, Uribe BI. Guías de práctica clínica basadas en la evidencia, Lupus Eritematoso Sistémico. Seguro social 2011 11-50.
- Abud-Mendoza C, et al. Treating severe systemic lupus erythematosus with rituximab. An open study. Reumatol Clin. 2009. doi:10.1016/J. Reuma. 2008.09.008
- Calderón SRE, et al. Tratamiento con rituximab para la trombocitopenia secundaria a lupus eritematoso sistémico. Reumatol Clin. 2009. doi:10.1016/j.reuma.2009.04.013.
- Calvo AJ, Mata C, Aurrecoechea E. Utilización de terapias hiperestrogénicas en el lupus eritematoso sistémico. Reumatol Clin. 2010;6(5):264–267
- Toong C, Adelstein S, Giang PT. Clearing the complexity: immune complexes and their treatment in lupus nephritis; International Journal of Nephrology and Renovascular Disease 2011:4 17–28.
- Romero ATB, García MGE. Citocinas y lupus eritematoso sistémico. Gac Méd Caracas 2009;117(3):196-211.
- Silva CE. Inmunopatogenia del Lupus Eritematoso Sistémico, Parte I: Factores Predisponentes y Eventos Iniciales. Rev. chil. reumatol. 2009; 25(3):108-113.
- Alarcon SG. Multiethnic lupus cohorts: What have they taught us?. Reumatol Clin. 2011;7(1):3–6
- Tsokos GC, Gordon C, Smolen JS, editors. Systemic Lupus Erythematosus. 2007.
- Otón T, et al. Terapia biológica en el lupus eritematoso sistémico. ¿Hay vida más allá del linfocito B?. Semin Fund Esp Reumatol. 2011. doi:10.1016/j.semreu.2010.08.001
- Estévez del Toro M, et al. Daño en pacientes cubanos con lupus eritematoso sistémico. Relación con características de la enfermedad. Reumatol Clin. 2009.doi:10.1016/j.reuma.2009.04.007.
- Santiago CWG, Morales RLN, Peña RDM, Chacón PNP. Lupus Eritematoso Sistémico Seronegativo. Repertorio de Medicina y Cirugía. 2010;19(1):52-56
- Ruiz VR. LUPUS ERITEMATOSO: Actualización clínico-diagnóstica y enfoque terapéutico. Dermathea innovación.
- Alarcón RME. Genética del lupus eritematoso generalizado. ¿Qué se sabe y a dónde se va?. Reumatol Clin.2010;6(1):1–2
- Vargas MJ, Moreira VC, Núñez DK, Núñez dm.causas de muerte en pacientes con lupus eritematoso sistémico (les) en el hospital méxico de 1989 a 2004. Revista médica de Costa Rica y Centro América LXVII (594) 2010 337-343.
- Andrade-Ortega L. Eficacia de rituximab comparado con ciclofosfamida en pacientes con manifestaciones graves de lupus eritematoso generalizado. Estudio aleatorizado y multicéntrico. Reumatol Clin.2010;6(5):250–255
- Silva CE. Inmunopatogenia del Lupus Eritematoso Sistémico, parte II: Rol de los Componentes del Sistema Inmune y de los Autoanticuerpos. Rev. chil. reumatol. 2009; 25(4):140-147
- Soto-Vargas J. Inmunopatogenia de Lupus Eritematoso Sistémico. Rev Med MD 2011; 2 (3) 170-179
- Guarnizo P, Vásquez G. Polimorfismo de citoquinas en lupus eritematoso sistémico. Revista Colombiana de Reumatología 2004 pp. 209-216
- Úcar AE, Rivera GN. Comorbilidad en lupus eritematoso sistémico. Reumatol Clin. 2008;4 Supl 1:S17-21
- Quintana LG, et al. Aplicación clínica de los anticuerpos en lupus eritematoso sistémico. Revista Colombiana de Reumatología. vol. 10 No. 1, marzo 2003 32-45.
- Sánchez AAI. Lupus eritematoso sistémico. Aspectos clínicos poco frecuentes. Reumatol Clin. 2008;4 Supl 1:S28-30
- Garcia-Consuera MJ. Lupus Eritematoso Sistémico. Protocolos diagnósticos y terapéuticos en pediatría. Reumatología sf. 59-64
- Cabiedes J, Nuñez ACA. Anticuerpos antinucleares. Reumatol Clin.2010;6(4):224–230
- Javier-Zepeda CA. Anticuerpos anti-nucleares Una familia diversa. Rev Med Hond 2002; 70:189-193
- Requilme L. Lupus Eritematoso.Dermatología: Correlación clínicopatológica 355-360.
- Hernández RDF, Cabiedes J. Técnicas inmunológicas que apoyan el diagnóstico de las enfermedades autoinmunes. Reumatol Clin.2010;6(3):173–177
- Eristian EE. The new approach in diagnostics of systemic lupus erythematosus the immunoagglutination of collaurin, Klin Lab Diagn 2001 Jul; (7):39-41.
- CRITERIOS DIAGNÓSTICOS DEL LUPUS ERITEMATOSO SISTÉMICO. Avances médicos 2004 1-2
- Benseler SM, Silverman ED. Lupus eritematoso sistémico. Pediatr Clin N Am 52 (2005) 443 – 467
- 35.Stringa O, Trojelli P. Consenso sobre diagnóstico y tratamiento de Lupus Eritematoso. Sociedad Argentina de dermatología 2006 2-34.

